5.1 Rekapitulation Schulwissen
Dass Carbonsäuren ihr Proton abgeben und beispielsweise mit Natrium Salze R-COO- Na+ bilden können, wird im Unterricht normalerweise behandelt. Im Zusammenhang mit organischen Reaktionsmechanismen wird sicherlich auch erwähnt, das Carboxylat-Ionen wie das Acetat-Ion CH3-COO- recht starke Nucleophile sind, also beispielsweise nucleophile Substitutionen oder nucleophile Additionen eingehen können.
Auch die Tatsache, dass die Carboxy-Gruppe reduziert werden kann, wird oft thematisiert, oft im Zusammenhang mit Oxidationszahlen. Aus Carbonsäuren können Aldehyde und primäre Alkohole hergestellt werden (Reduktion um zwei bzw. vier Stufen).
Im Biologie-Unterricht ist wahrscheinlich auch der Begriff Decarboxylierung vorgekommen, vor allem bei der Behandlung des Citratzyklus. Aus einem C6-Körper (Citrat) wird so in mehreren Schritten, die zwei Decarboxylierungen (Abspaltung von CO2) einschließen, ein C4-Körper (Oxalacetat), der wieder einen C2-Körper anlagert (Acetyl-CoA) und dann erneut zum C6-Körper (Citrat) wird.
Das Thema "Citratzyklus" nimmt eine ganze Reihe von Seiten auf dieser Homepage in Anspruch. Sie können sich dort gerne über die Einzelheiten informieren, wenn Sie mehr dazu wissen möchten (oder müssen).
Die nucleophile Substitution der Carboxy-OH-Gruppe wird dagegen im Schulunterricht meistens nicht behandelt. Auf diese Weise können aus Carbonsäuren nämlich Carbonsäure-Chloride, -Anhydrate und -Amide entstehen. Auch die Bildung von Carbonsäure-Estern erfolgt durch die nucleophile Substitution der OH-Gruppe gegen einen Alkohol-Rest. Von der säurekatalysierten Veresterung einer Carbonsäure haben Sie im Unterricht wahrscheinlich schon einmal etwas gehört.
Dieses Thema wird normalerweise im Chemieunterricht der Oberstufe behandelt. Gehen Sie auf diese Seite, wenn Sie Ihre Kenntnisse noch einmal rekapitulieren wollen. Allerdings wird diese Thema auch im Abschnitt 5.2 (Studienvorbereitung) noch einmal behandelt.
5.2 Studienvorbereitung
Auch diese Seite ist wieder einmal viel länger geworden als ursprünglich geplant. Daher ist es sinnvoll, eine kleine Gliederung an den Anfang dieses Abschnitts zu stellen.
5.2.1 Die Carboxy-Gruppe
Wenn wir die Reaktionen der Carbonsäuren besprechen wollen, sollten wir uns zunächst einmal die Struktur der Carboxy-Gruppe näher ansehen:

Chemische Eigenschaften der Carboxygruppe
Autor: Ulrich Helmich 05/2024, Lizenz: Public domain
Die Schemazeichnung zeigt mehrere Reaktionsmöglichkeiten auf.
- Die C=O-Doppelbindung der COOH-Gruppe kann - zumindest theoretisch - Additionsreaktionen eingehen, ähnlich wie bei Aldehyden oder Ketonen (nucleophile Addition).
- Die OH-Gruppe sitzt an einem positiv polarisierten C-Atom und kann leicht als Nucleofug abgespalten und durch ein anderes Nucleophil substituiert werden (nucleophile Substitution).
- Das alpha-C-Atom ist ebenfalls noch etwas positiv polarisiert und kann daher auch von Nucleophilen angegriffen werden.
- Das H-Atom der OH-Gruppe kann als Proton abgespalten werden (Säure-Base-Reaktion).
- Nach der Abspaltung des Protons kann das verbliebene Carboxylat-Anion R-COO- selbst als Nucleophil auftreten.
Der fünfte Punkt ist in der Schemazeichnung noch nicht berücksichtigt worden, weil es sich ja nicht um eine Reaktion der Carboxy-Gruppe handelt, sondern um eine Reaktion des Carboxylat-Ions.
Blick in die Fachliteratur:
Ein Blick in die Fachliteratur zeigt aber, dass fast ausschließlich die Nucleophile Substitution ausführlich behandelt wird; in den Chemie-Vorlesungen wird das wahrscheinlich ähnlich sein.
Die "Zusammenfassung der Reaktionen der Carboxylgruppe" in [2] listet folgende Reaktionen auf:
- Reaktion mit OH- zum Carboxylat-Ion
- Reaktionen mit R-OH zu Estern
- Reaktionen mit SOCl2 zu Carbonsäurechloriden
- Reaktion mit LiAlH4 zu primären Alkoholen
- Decarboxylierung zu Alkanen
Wir beginnen unsere Betrachtungen nun mit den Substitutionsreaktionen.
5.2.2 Substitutionsreaktionen ↑
Schauen wir uns dazu die folgende Abbildung an:
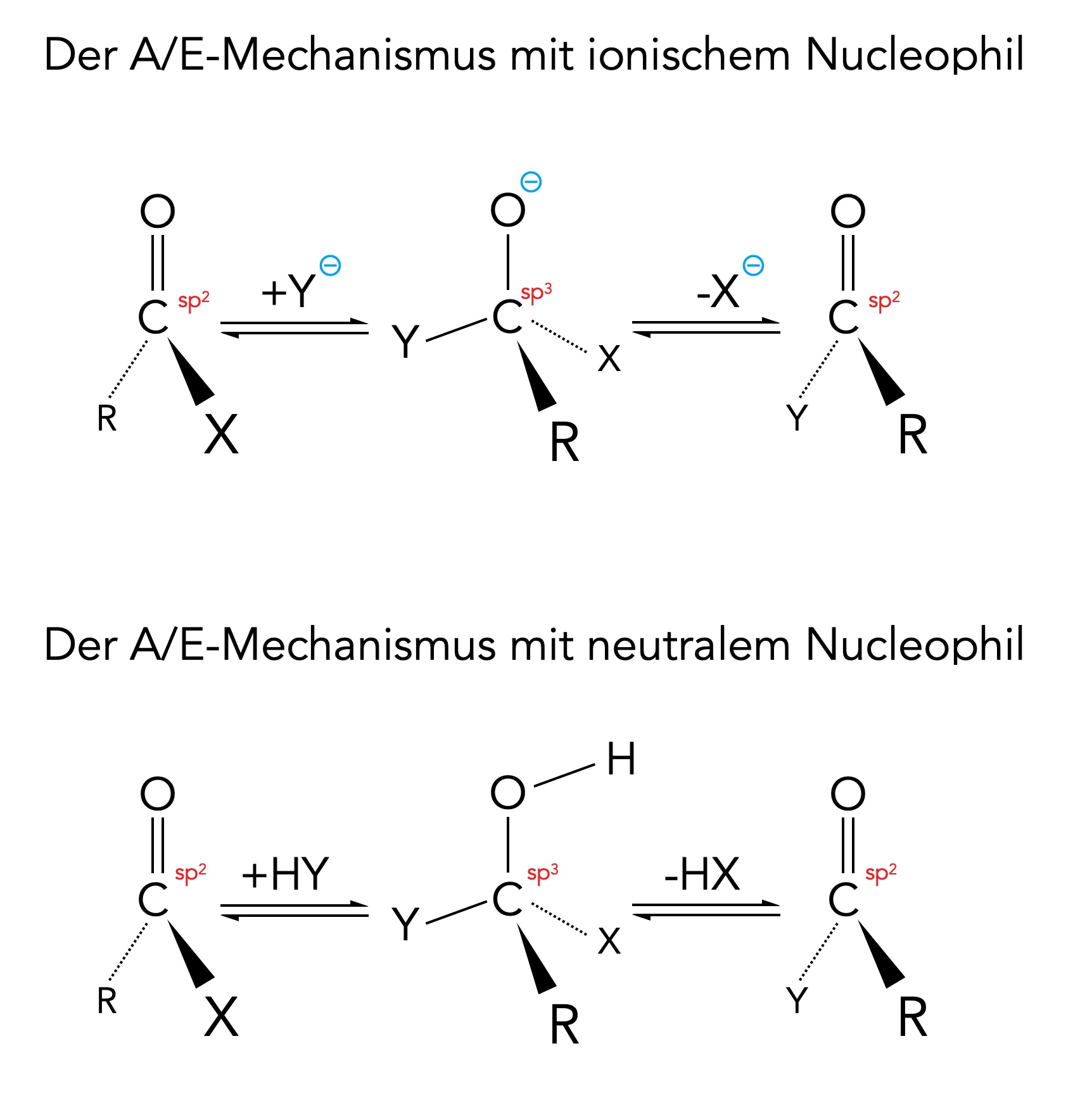
Der Additions-Eliminierungs-Mechanismus bei Carbonsäure-Derivaten R-COX
Autor: Ulrich Helmich 06/2024, Lizenz: Public domain
Schritt 1
Im ersten Reaktionsschritt (A für Addition) setzt sich ein Nucleophil Y:- oder HY an das positiv polarisierte C-Atom der COOH-Gruppe. Die mitgebrachte negative Ladung wird dabei von dem Carbonyl-O-Atom übernommen. Das C-Atom wechselt bei dieser Addition seinen Hybridisierungszustand von sp2 nach sp3.
Schritt 2
Theoretisch könnte sich nun - ähnlich wie bei der Reaktion der Aldehyde oder Ketone - ein Elektrophil an das negative Sauerstoff-Atom setzen. Bei Carbonsäuren und Carbonsäure-Derivaten findet aber fast ausschließlich die Eliminierung des Nucleofugs X statt, entweder als X- oder als HX.
Der Additions-Eliminierungs-Mechanismus A/E
Allerdings verläuft dieser Substitutionsmechanismus völlig anders, als wir es von der bekannten SN1- oder SN2-Reaktion kennen.
Bei der SN1 findet im ersten Schritt eine Eliminierung statt und nicht eine Addition, wie bei dem A/E-Mechanismus. Und bei der SN2 verlaufen Addition und Eliminierung gleichzeitig und nicht in zwei Schritten wie beim A/E-Mechanismus.
Hier ein kurzer Vergleich:
| Schritt | SN1 | SN2 | A/E |
| 1 | Eliminierung von X | Addition und Eliminierung zeitgleich | Addition von Y |
| 2 | Addition von Y | Eliminierung von X | |
| Zwischenstufe | planares Carbeniumion sp2 |
pentavalenter Übergangszustand sp2 |
tetraedrisches Anion sp3 |
Auf dieser Seite im Chemie-Lexikon ist dieser neue Reaktionsmechanismus ausführlich beschrieben. Zunächst wird er mit den SN1- und dem SN2-Mechanismus verglichen, dann wird der Grundmechanismus - ähnlich wie auf dieser Seite - sowie der basenkatalysierte und der säurekatalysierte A/E-Mechanismus Schritt für Schritt vorgestellt und erläutert.
5.2.2.1 Veresterung ↑
Carbonsäure-Ester sind Verbindungen R-CO-O-R', die sich formal aus einer Carbonsäure R-COOH und einem Alkohol R'-OH zusammensetzen.
Gebildet werden Ester durch Kondensation einer Carbonsäure mit einem Alkohol:
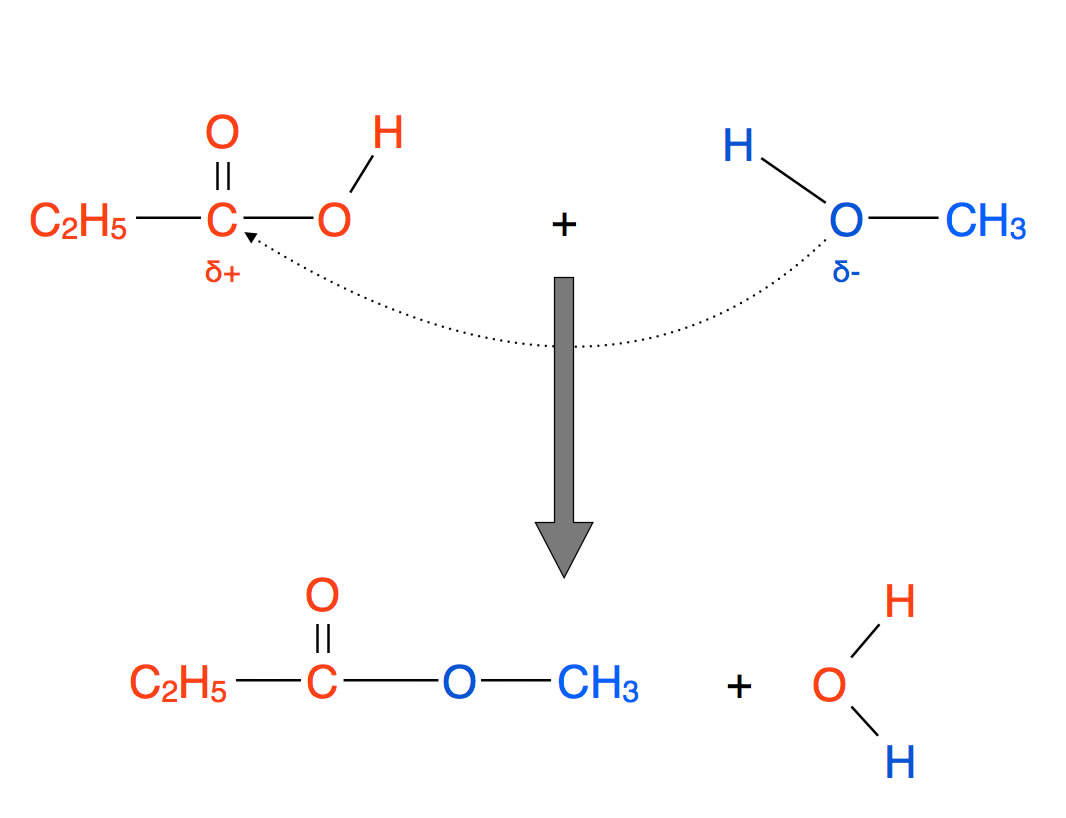
Bildung eines Esters aus Propionsäure und Methanol
Autor: Ulrich Helmich 2022, Lizenz: Public domain
Andere Synthese-Möglichkeiten sind die Reaktion eines Carbonsäure-Chlorids mit einem Alkohol unter Freisetzung von HCl (statt H2O) oder die Umsetzung eines Carbonsäure-Anhydrids mit zwei Molekülen Alkohol unter Freisetzung von zwei Molekülen H2O.
Führt man die Veresterung von Essigsäure mit Ethanol ohne Zusatz einer Säure durch, so läuft die Reaktion extrem langsam ab. Bis zur Einstellung eines chemischen Gleichgewichts können Monate bis Jahre vergehen. Durch Zusatz weniger Tropfen Schwefelsäure kann man diesen Vorgang aber erheblich beschleunigen. Schauen wir uns den Mechanismus der säurekatalysierten Veresterung jetzt einmal etwas näher an.
Die säurekatalysierte Veresterung läuft nach dem A/E-Mechanismus ab
Um deutlich zu machen, dass diese Reaktion tatsächlich nach dem A/E-Mechanismus abläuft, habe ich einfach die Graphik, die ich für den säurekatalysierten A/E-Mechanismus erstellt habe, so abgewandelt, dass sie die säurekatalysierte Esterbildung aus einer Carbonsäure und einem Alkohol zeigt.
Eine ausführliche Schritt-für-Schritt-Darstellung dieses Mechanismus finden Sie auf der Vertiefungsseite in dieser Abteilung.
5.2.2.2 Bildung von Carbonsäure-Chloriden ↑
Die Synthese eines Carbonsäure-Chlorids wird in der Regel mit Hilfe der Verbindung Thionylchlorid durchgeführt:
R-COOH + SOCl2 → R-CO-Cl + SO2 + HCl
Alternative Methoden zur Bildung von Carbonsäure-Chloriden aus Carbonsäuren sind [4]:
- die Umsetzung mit Phosgen COCl2 unter Abspaltung von CO2 und HCl,
- die Umsetzung mit Phosphor(III)-chlorid PCl3 unter Abspaltung von Phosphonsäure H3PO3,
- die Reaktion mit Phosphor(V)-chlorid PCl5 unter Bildung von Phosphoroxychlorid POCl3 und HCl.
Auf dieser Vertiefungsseite wird die Darstellung von Carbonsäure-Chloriden aus Carbonsäuren mit Hilfe der Verbindung Thionylchlorid ausführlich dargestellt.
Diese Lexikon-Seite enthält weitere Informationen zum Thema "Carbonsäure-Chloride".
Aktivierte Carbonsäuren
Carbonsäure-Chloride werden auf als aktivierte Carbonsäuren bezeichnet, weil sie im Prinzip die gleichen Reaktionen wie eine Carbonsäure eingehen, dabei aber wesentlich reaktiver sind. Die Veresterung einer Carbonsäure kann beispielsweise sehr lange dauern, ohne Katalysator benötigen manche Carbonsäuren mehrere Monate, bis sich ein chemisches Gleichgewicht eingestellt hat. Lässt man den Alkohol dagegen mit dem Carbonsäure-Chlorid reagieren, bildet sich der gleiche Ester quasi "in Null komma Nichts". Allerdings wird dann kein Wasser als Nebenprodukt freigesetzt, sondern Chlorwasserstoff.
5.2.2.3 Bildung von Carbonsäure-Amiden ↑
Die Stoffklassen der Amide und der Amine werden gern verwechselt, daher wollen wir hier zunächst einmal die beiden Klassen gegeneinander abgrenzen.
Amine sind Derivate des Ammoniaks NH3. Bei den primären Aminen ist eines der drei H-Atome durch eine Alkyl-Gruppe ersetzt, bei sekundären Aminen sind zwei H-Atome ersetzt, und bei tertiären Aminen sogar alle drei H-Atome.
Amide können ebenfalls als Derivate des Ammoniaks angesehen werden. Allerdings werden hier die H-Atom nicht durch einfach Alkyl-Reste ersetzt, sondern durch Säure-Reste - vor allem durch Carbonsäure-Reste.
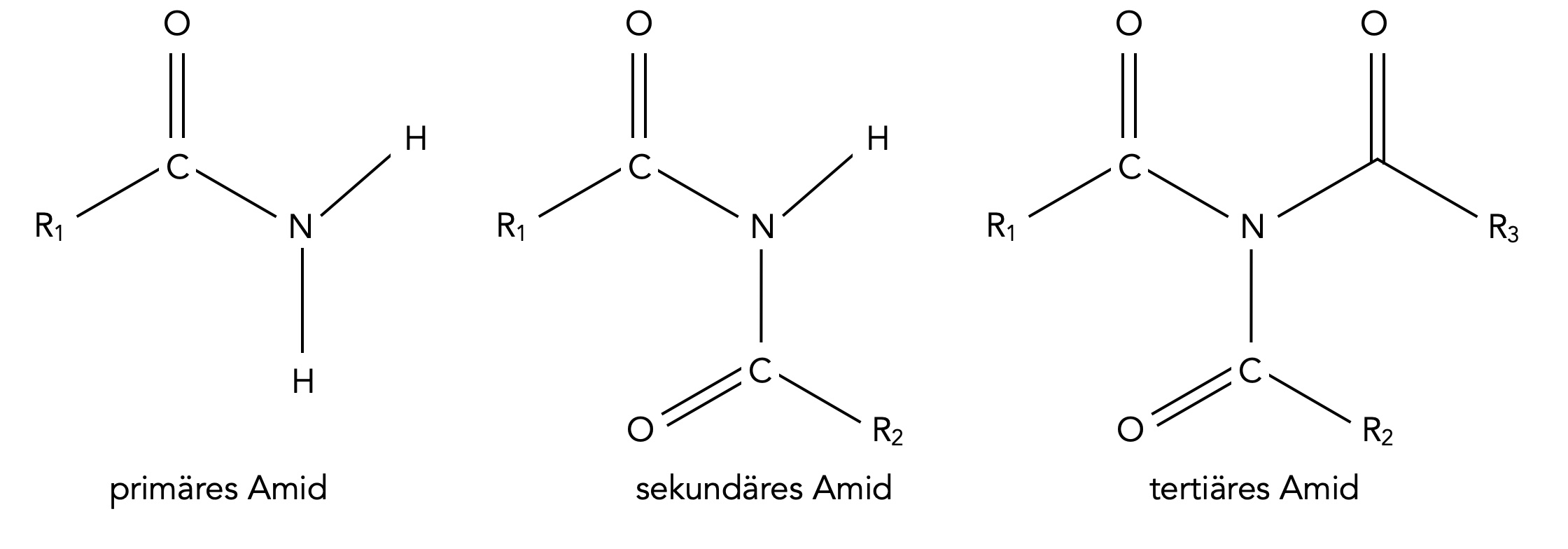
Primäre, sekundäre und tertiäre Amide des Ammoniaks
Autor: Ulrich Helmich 03/2024, Lizenz: Public domain
Bei primären Amiden wurde eines der drei H-Atom des Ammoniaks durch einen Acyl-Rest ersetzt, bei sekundären Amiden finden wir zwei Acyl-Reste und nur ein H-Atom, und bei tertiären Amiden sind alle drei H-Atome durch Acyl-Reste ersetzt worden.
Man kann die Carbonsäure-Amide aber auch aus einem anderen Blickwinkel betrachten, nicht als Derivate des Ammoniaks, sondern als Derivate der Carbonsäuren. Unter diesem Gesichtspunkt würde man ein Amid definieren als eine Carbonsäure, deren OH-Gruppe durch eine Amino- oder Amin-Gruppe ersetzt wurde.
Synthese
Theoretisch könnte man ein Carbonsäure-Amid herstellen, indem man einfach eine Carbonsäure mit Ammoniak oder einem Amin zusammen gibt. Leider erhält man so kein Amid, sondern ein Ammoniumsalz. Denn die Carbonsäure kann ja nicht nur nucleophile Substitutionen eingehen, sondern auch als Brönsted-Säure ein Proton abgeben. Und genau das würde passieren, wenn man Essigsäure oder eine andere Carbonsäure mit Ammoniak oder einem primären Amin reagieren lässt. Die Stickstoff-Verbindung würde das Proton aufnehmen, und es würde ein Ammoniumsalz wie Ammoniumacetat entstehen, aber kein Carbonsäure-Amid.
Damit eine Carbonsäure ein Amid bilden kann, muss man die Carbonsäure zuvor aktivieren. Das geschieht in der Regel durch die Überführung der Carbonsäure in das entsprechende Carbonsäure-Chlorid.
Auf dieser Vertiefungsseite finden Sie weitere Informationen zur Bildung von Carbonsäure-Amiden.
5.2.2.4 Carbonsäure-Anhydride ↑
Rein formal sind Carbonsäure-Anhydride Verbindungen, die aus zwei Molekülen einer Carbonsäure bestehen, deren OH-Gruppen sich unter Abspaltung von Wasser vereinigt haben (Kondensation).
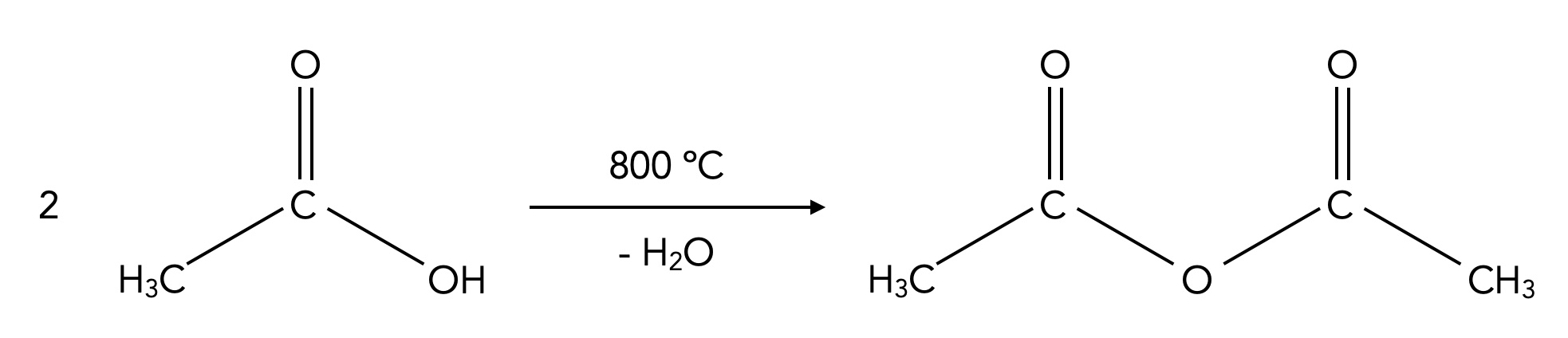
Industrielle Herstellung von Essigsäureanhydrid
Autor: Ulrich Helmich 06/2024, Lizenz: Public domain
Hier sehen wir ein Verfahren zur industriellen Herstellung von Essigsäureanhydrid, einer wichtigen Grundchemikalie der chemischen Industrie.
Allerdings reagieren die Carbonsäuren selbst nur recht schwer zu Anhydriden, das Carboxyl-C-Atom ist kein besonders starkes Elektrophil bzw. Angriffsziel für Nucleophile. Carbonsäure-Chloride dagegen haben wegen des -I-Effekts des Cl-Atoms ein viel stärker positiv polarisiertes Carbonyl-C-Atom und werden daher viel leichter von Nucleophilen angegriffen. Daher reagiert ein Carbonsäure-Chlorid mit einer Carbonsäure recht schnell zu einem Carbonsäure-Anhydrid.
Auf dieser Vertiefungsseite wird die Bildung von Carbonsäure-Anhydriden aus Carbonsäuren und Carbonsäure-Chloriden näher besprochen.
5.2.3 Reduktion zu Alkoholen ↑
Alkohole können meistens recht leicht aus anderen Verbindungen synthetisiert werden, beispielsweise aus Alkenen durch Addition von Wasser oder aus Halogenalkanen durch Substitution des Halogen-Atoms durch eine OH-Gruppe. Eine andere wichtige Methode zur Herstellung bestimmter Alkohole ist die Reduktion von Carbonylverbindungen und Carbonsäuren.
Das beliebteste Reduktionsmittel, mit dem man aus einer Carbonsäure einen primären Alkohol herstellen kann, ist das Lithiumaluminiumhydrid LiAlH4[5,6]. Der genaue Verlauf der Reduktion ist hier jetzt nicht relevant, auf jeden Fall entsteht dann unter recht milden Bedingungen der passende primäre Alkohol in hoher Ausbeute aus der Carbonsäure. Allerdings ist das Lithiumaluminiumhydrid recht teuer, daher wird es nur bei der Synthese von Arzneimitteln oder Hormonen eingesetzt[5].
Hier ein möglicher Mechanismus dieser Reduktion in vier Schritten:

Die Gesamtgleichung dieser Umsetzung wäre dann:
CH3-COOH + 2 AlH4- + H3O+ → CH3-CH2-OH + 2 AlH3 + OH- + H2O
Im berühmten Organikum [8] lesen wir zu dieser Reaktion:
"... sind gewisse Metallhydride H-M in der Lage, als H-Nucleophile zu wirken und ein Wasserstoffatom mit seinen Bindungselektronen als Hydridion auf das C-Atom der Carbonylgruppe zu übertragen. Das Hydridion tritt dabei aber nicht frei auf, sondern reagiert unter konzertierter Spaltung der M-H-Bindung und Knüpfung der C-H-Bindung."
Auf dieser Vertiefungsseite werden verschiedene Versionen des Mechanismus aus der Fachliteratur und aus dem Internet vergleichend gegenübergestellt.
5.2.4 Decarboxylierungen ↑
Grundprinzip
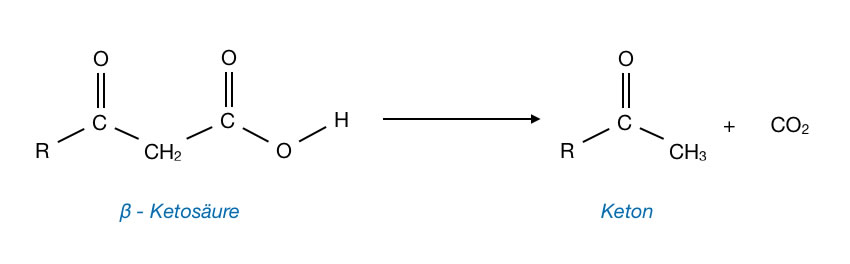
Unter einer Decarboxylierung versteht man die Abspaltung der COOH-Gruppe einer Carbonsäure als Kohlendioxid CO2.
Die Oxidationszahl des alpha-C-Atoms ändert sich dadurch leicht von -II zu -III, was der Reduktion um eine Stufe entspricht:
Änderung der Oxidationszahlen durch eine Decarboxylierung
Autor: Ulrich Helmich 06/2024, Lizenz: Public domain
Die Oxidationszahl des Carboxy-C-Atoms steigt dagegen von +III auf +IV.
Decarboxylierungen kommen vor allem bei Ketosäuren vor.
Normale Carbonsäuren können nur bei recht hohen Temperaturen decarboxyliert werden, während Ketosäuren oder Dicarbonsäuren schon bei relativ niedrigen Temperaturen zwischen 100 und 150 ºC decarboxyliert werden.
Ein bekanntes und in der Industrie genutztes Verfahren zur Decarboxylierung von Carbonsäure-Salzen ist die Kolbe-Elektrolyse.
Am Pluspol wird dem Carboxylat-Anion ein Elektron entzogen, es entsteht dann ein Carboxylat-Radikal mit einem einsamen Elektron an dem O-Atom.
Unter Abspaltung von CO2 entsteht dann ein Alkyl-Radikal, von denen sich je zwei zu einem Alkan-Molekül vereinigen.
Auf dieser Vertiefungsseite erfahren Sie Einzelheiten zum Ablauf der Decarboxylierung, auch die Kolbe-Elektrolyse wird näher erläutert.
Decarboxylierungen im Citratzyklus
Decarboxylierungen finden in jeder Zelle des menschlichen Körpers viele tausend Mal pro Sekunde statt, und zwar im Citratzyklus (Zitronensäurezyklus). Hier ein ganz kurzer und grober Überblick über diesen wichtigen Stoffwechselprozess, der in fast allen Lebewesen ständig zur Energiegewinnung abläuft. Wussten Sie, dass das CO2, das wir ausatmen, aus den Decarboxylierungen kommt, die im Citratzyklus stattfinden?
Wie bereits in der Einleitung gesagt, ist das wohl bekannteste Beispiel für eine Decarboxylierung die Umwandlung von Zitronensäure in einen C5-Körper und dann in einen C4-Körper.
Der Citratzyklus im Überblick
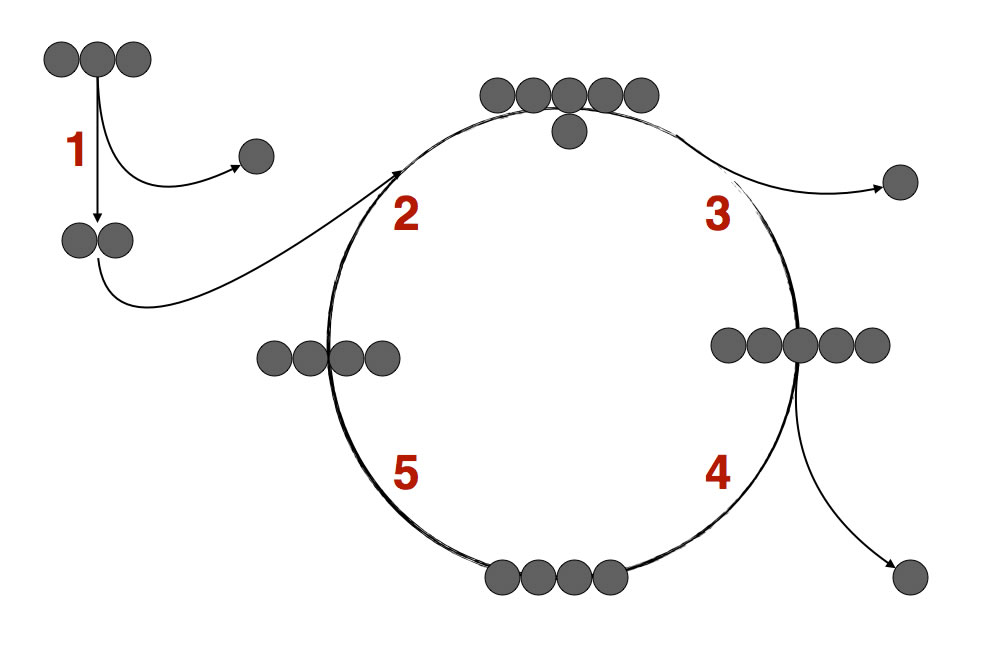
Das Grundprinzip des Citratzyklus
Autor: Ulrich Helmich 2015, Lizenz: Public domain
Hier sehen wir das Grundprinzip des Citratzyklus, auf das Wesentliche beschränkt. Oben links kommt ein Pyruvat-Molekül aus der Glycolyse an, es enthält drei Kohlenstoff-Atome, daher wurde das Pyruvat hier durch drei graue Kreise dargestellt.
Erste Decarboxylierung:
Das Pyruvat wird decarboxyliert, es entsteht ein C2-Körper, der ähnlich wie ein Essigsäure-Molekül aufgebaut ist. Das Coenzym A überträgt diesen C2-Körper dann als Acetyl-CoA in den eigentlichen Citratzyklus.
Der Acetyl-Rest (C2) verbindet sich nun mit einem Oxalacetat-Molekül (C4) zur Zitronensäure (C6) bzw. zum Citrat, dem Anion der Zitronensäure.
Zweite Decarboxylierung:
Im nächsten Schritt (3) wird Kohlendioxid aus dem Citrat abgespalten, und es bleibt ein C5-Molekül zurück.
Dritte Decarboxylierung:
Aus diesem C5-Molekül wird noch mal ein Kohlendioxid abgespalten (4), so dass ein C4-Molekül zurück bleibt.
Weitere Schritte:
Dieses C4-Molekül wird etwas umgewandelt, bleibt dabei aber ein C4-Molekül. Dieser Schritt wird unten links in dem Zyklus angedeutet (5).
Das veränderte C4-Molekül schließlich verbindet sich wieder mit einem neuen C2-Molekül (2), und der Zyklus ist geschlossen. Es entsteht wieder ein Citrat-Molekül, und alles geht von vorn los.
Diese Vertiefungsseite beschäftigt sich eingehend mit den drei oben genannten Decarboxylierungen.
5.2.5. Verlängerung um eine CH2-Gruppe
In seiner Fortgeschrittenen-Vorlesung "Organische Chemie 2.34" stellt Prof. Dyker seinen Studierenden folgende Denkaufgabe:
Aufgabe
Wie kann man die folgende Carbonsäure um eine Methylgruppe verlängern:
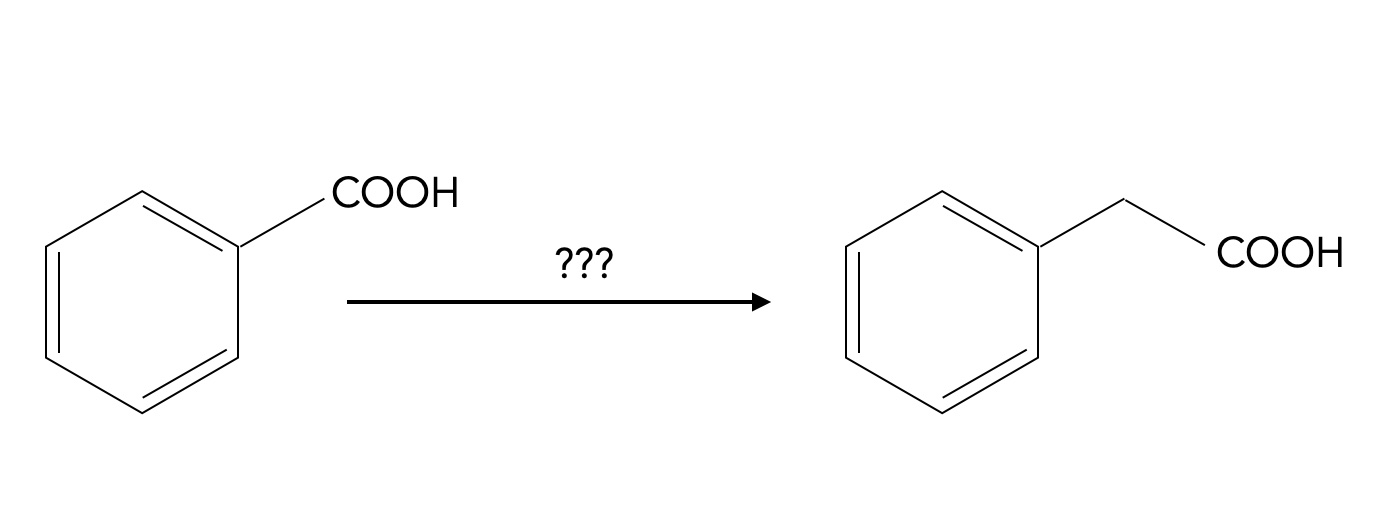
Lösung
Diejenigen von Ihnen, die aus dem Schulunterricht die Unterrichtsreihe "Vom Alkan zum Plexiglas" kennengelernt haben, sollten diese Aufgabe lösen können. Falls nicht, können Sie sich auf dieser Homepage auf der Seite "Vom Propan zum Plexiglas" informieren.
Die Reaktionsfolge zur Synthese der verlängerten Carbonsäure sieht nach Prof. Dyker folgendermaßen aus:
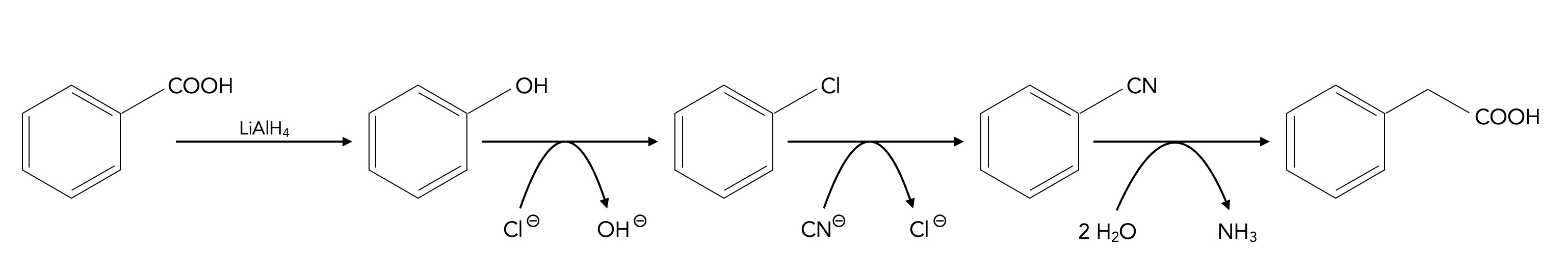
Zunächst wird die Carbonsäure zum Alkohol reduziert, das geschieht mittels Lithiumaluminiumhydrid LiAlH4. In einer Nucleophilen Substitution wird dann die OH-Gruppe durch ein Halogen-Atom ersetzt. Eine zweite Nucleophile Substitution tauscht dann das Halogen-Atom gegen eine Cyanid-Gruppe aus. Die Hydrolyse der CN-Gruppe schließlich liefert die gewünschte Carbonsäure mit einem C-Atom mehr als die Ausgangssubstanz.
5.2.6 Reaktionen am α-C-Atom
Starke Basen entziehen einer Carbonsäure nicht nur das H-Atom der Carboxygruppe, sondern darüber hinaus auch noch ein H-Atom des α-C-Atoms:
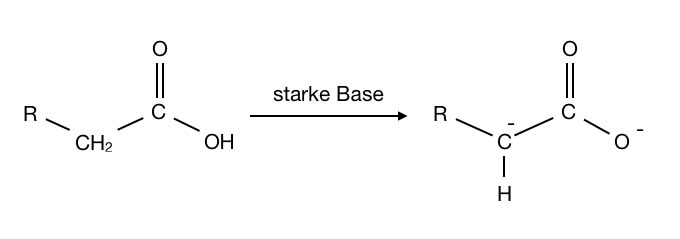
Eine starke Base kann einer Carbonsäure ein α-H-Atom entreißen
Autor: Ulrich Helmich 05/2023, Lizenz: Public domain
An das negativ geladene C-Atom kann nun leicht ein elektrophiles Carbenium-Ion angreifen, wie sie zum Beispiel beim ersten Schritt der SN1-Reaktion entstehen oder bei der Addition eines Protons an die C=C-Doppelbindung.
Die Hell-Volhard-Zelinsky-Reaktion
Bei der Hell-Volhard-Zelinsky-Reaktion wird eine Carbonsäure in α-Stellung monobromiert. Hier die einzelnen Schritte dieser Namensreaktion (nach [1]).
1. Durch Einwirkung von PBr3 auf eine Carbonsäure bildet sich ein Acylbromid:
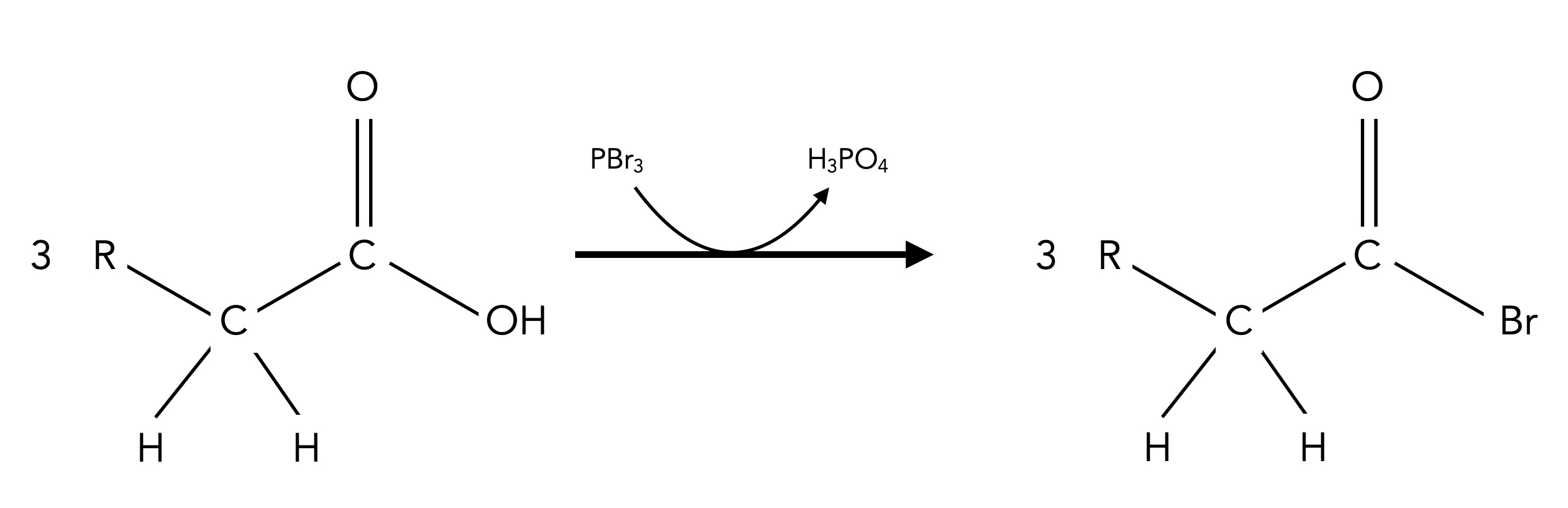
Schritt 1 der Hell-Volhard-Zelinsky-Reaktion
Autor: Ulrich Helmich 06/2024, Lizenz: Public domain
2. Aus der Keto-Form bildet sich schnell die Enolform:
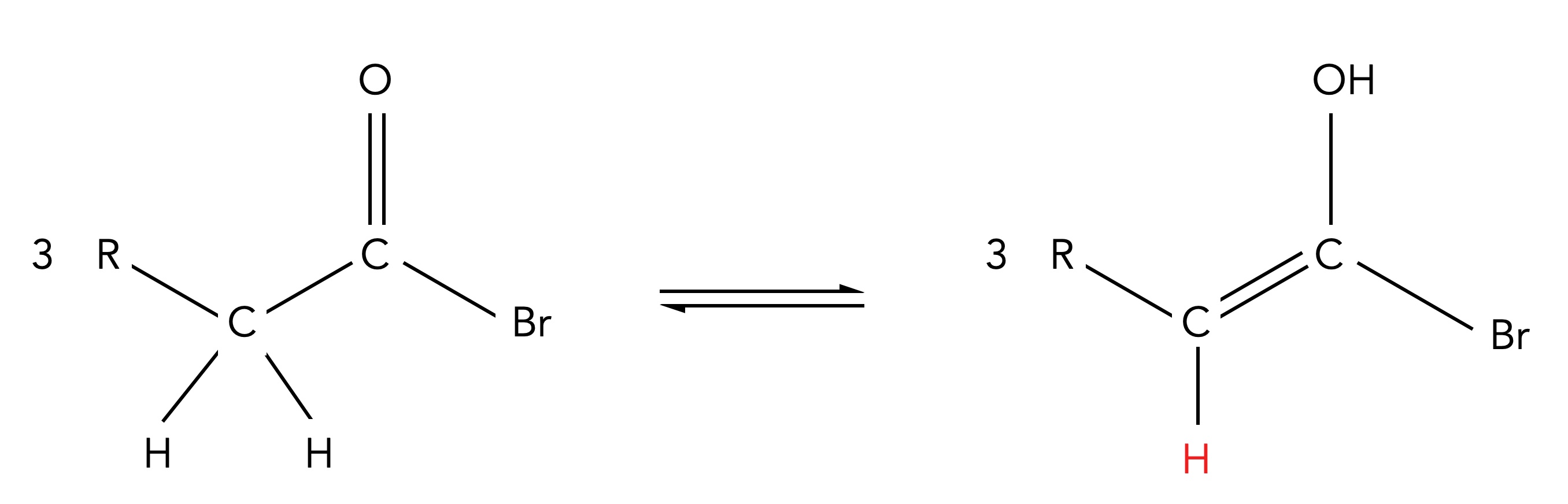
Schritt 2 der Hell-Volhard-Zelinsky-Reaktion
Autor: Ulrich Helmich 06/2024, Lizenz: Public domain
3. Diese Enolform kann dann leicht bromiert werden:
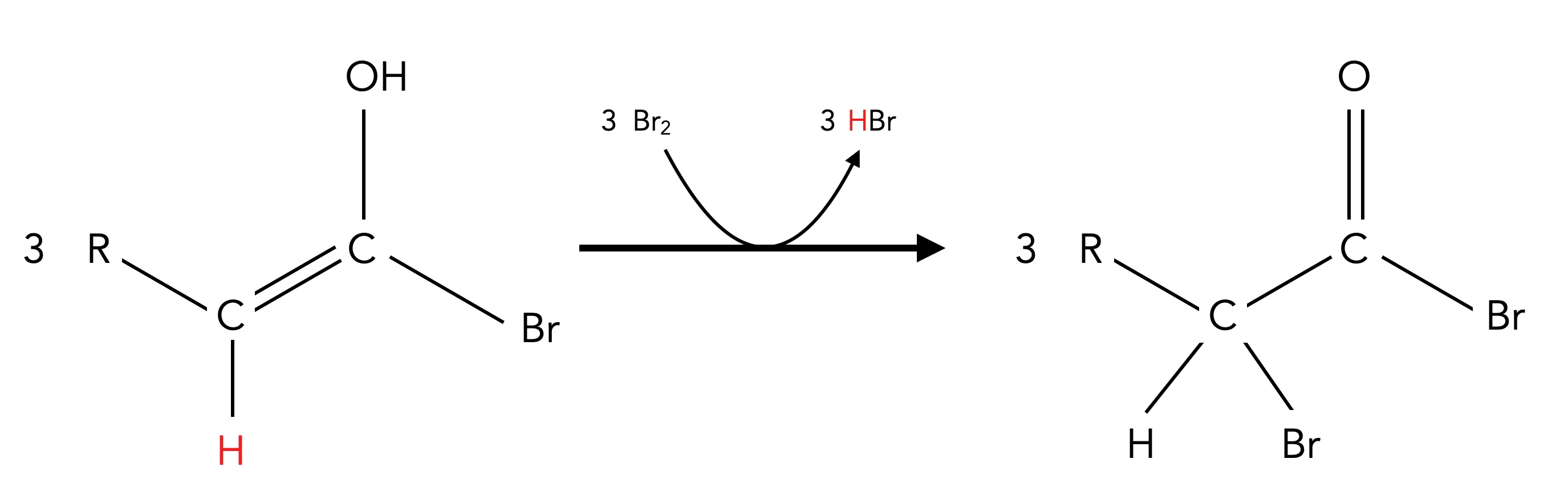
Schritt 3 der Hell-Volhard-Zelinsky-Reaktion
Autor: Ulrich Helmich 06/2024, Lizenz: Public domain
4. Es folgt nun eine sogenannte Austauschreaktion:
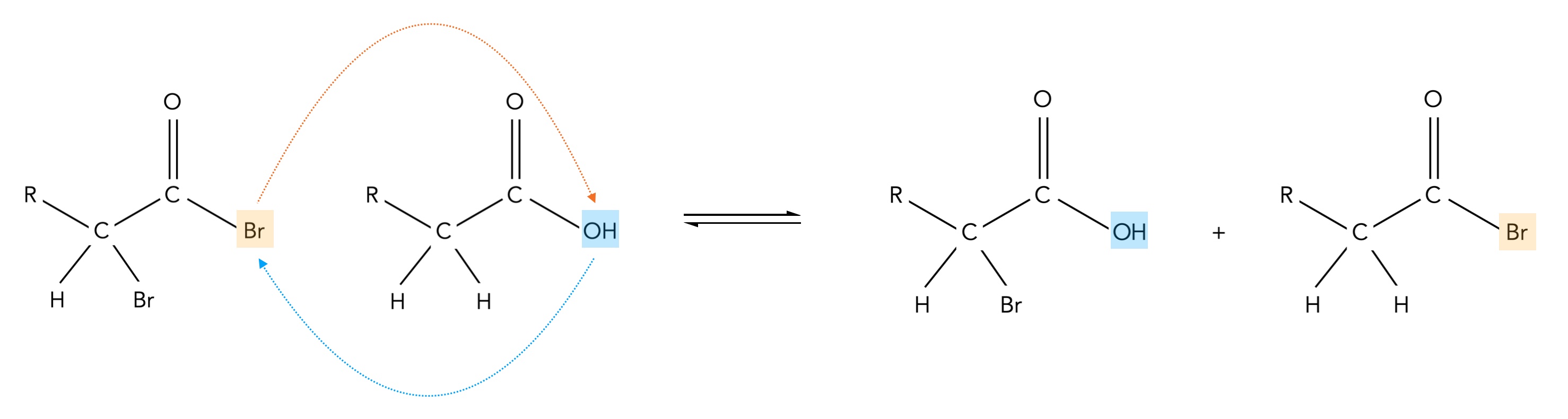
Schritt 4 der Hell-Volhard-Zelinsky-Reaktion
Autor: Ulrich Helmich 06/2024, Lizenz: Public domain
Das linke der beiden Reaktionsprodukte ist das gewünschte Endprodukt, die an alpha-Position monobromierte Carbonsäure. Das rechte der beiden Produkte geht erneut den Schritt 2 der Reaktion ein.
1. - 4. Gesamtdarstellung:
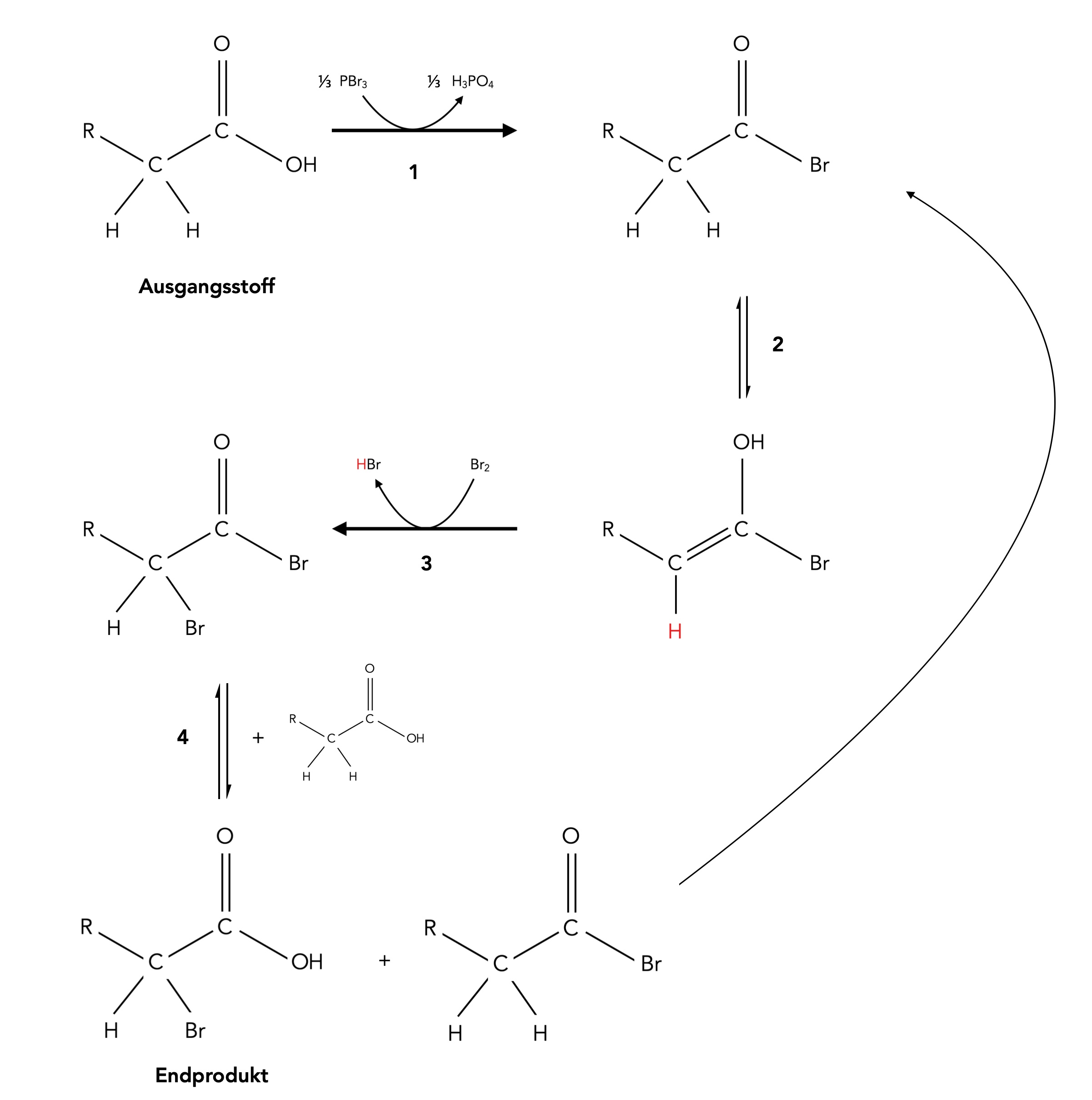
Gesamtdarstellung der Hell-Volhard-Zelinsky-Reaktion
Autor: Ulrich Helmich 06/2024, Lizenz: Public domain
In der engl. Wikipedia wird diese Reaktion noch ausführlicher erläutert.
5.2.7 Substitution der Carboxygruppe
Bei der Decarboxylierung wird die gesamte Carboxygruppe als CO2 abgespalten. Man kann die Carboxygruppe aber auch durch eine andere funktionelle Gruppe ersetzen. Bei der Hunsdiecker-Reaktion wird die COOH-Gruppe zum Beispiel durch ein Halogen-Atom ersetzt. Aus der Carbonsäure erhält man so ein Alkylhalogenid bzw. Halogenalkan.
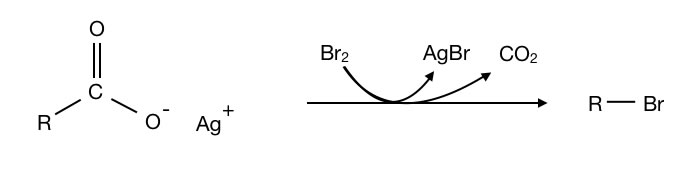
Die Hunsdiecker-Reaktion
Autor: Ulrich Helmich 05/2023, Lizenz: Public domain
Die Hunsdiecker-Reaktion ist allerdings ein reines Labor-Verfahren, in der Industrie wird sie nicht angewendet, weil zu viele Schadstoffe wie AgBr dabei entstehen.
Quellen:
- VollhardT, Schore: Organische Chemie. 6. Auflage, Weinheim 2020.
- Buddrus, Schmidt, Grundlagen der Organischen Chemie, 5. Auflage, De Gruyter-Verlag 2014.
- engl. Wikipedia, Artikel "Carboxylic acid"
- Wikipedia, Artikel "Carbonsäurechloride, Darstellung"
- Morrison, Boyd, Bhattacharjee: Organic Chemistry. 7. Auflage, Dorling Kindersley 2011.
- Chemgapedia, Artikel "Reaktion von Carbonsäuren" (nicht mehr zugänglich!)
- Carey, Sundberg: Organische Chemie, ein weiterführendes Lehrbuch. Weinheim 1995.
- Organikum, 22. Auflage, Weinheim 2004.