Allgemeines, Historisches
Der Begriff "Katalyse" ist schon sehr alt. Er wurde im Jahre 1835 von Berzelius geprägt. Nach Berzelius sind Katalysatoren "Stoffe, die durch ihre bloße Gegenwart chemische Reaktionen hervorrufen, welche sonst nicht stattfinden würden."
Wilhelm Ostwald fand dann 1894 bis 1901 heraus, dass ein Katalysator nur die Geschwindigkeit einer chemischen Reaktion beschleunigt, sich aber nicht auf die Lage des chemischen Gleichgewicht auswirkt. Ostwald definierte den Begriff "Katalysator" wie folgt: "Ein Katalysator ist ein Stoff, der die Geschwindigkeit einer Reaktion verändert, selbst jedoch nicht zu den Produkten dieser Reaktion gehört." [1]
Aber auch die Definition von Ostwald ist nach heutigem Kenntnisstand nicht vollständig. Eine ganze Reihe von chemischen Reaktionen können ohne einen Katalysator überhaupt nicht stattfinden. Dazu gehören viele biochemische Reaktionen in lebenden Zellen, aber auch viele industriell wichtige Polymerisierungsreaktionen.
Was in der Ostwald-Definition ebenfalls noch nicht erwähnt wird, ist die mitunter sehr hohe Spezifität vieler Katalysatoren. Gerade Enzyme (Biokatalysatoren) sind extrem spezifisch, was die Wahl ihrer Substrate angeht. Enzyme arbeiten nach dem Schlüssel-Schloss-Prinzip, das umzusetzende Substrat muss exakt in das aktive Zentrum hineinpassen. Ist das nicht der Fall, läuft die Katalyse mit verminderter Geschwindigkeit oder gar nicht ab. Diese Spezifität geht sogar so weit, dass das Enantiomer (Spiegelbild) eines Substrates nicht erkannt wird.
Alles über Enzyme erfahren Sie auf der entsprechenden Seite in meinem Biologie-Lexikon.
Nach neueren Erkenntnissen können Katalysatoren nicht nur die Geschwindigkeit einer Reaktion erhöhen, sondern auch Einfluss auf den Verlauf der Reaktion nehmen. Es gibt Fälle, in denen man aus den gleichen Edukten mit verschiedenen Katalysatoren unterschiedliche Produkte erhält. Propen beispielsweise kann mit den sogenannten Friedel-Crafts-Katalysatoren zu einem Gemisch oligomerer Olefine (Alkene, Cycloalkene, Polyene) umgesetzt werden, mit Ziegler-Natta-Katalysatoren dagegen bildet sich Polypropen [1, S. 485].
Katalysierte Reaktionen
Katalysierte Reaktionen verlaufen schneller als nicht-katalysierte Reaktionen, weil der Katalysator einen alternativen Reaktionspfad bewirkt, bei dem der geschwindigkeitsbestimmende Schritt eine deutlich geringere Aktivierungsenergie besitzt.
Ein Katalysator kann jedoch nicht den Betrag der freien Energie ΔG einer Reaktion verändern, und da die Lage des chemischen Gleichgewicht von ΔG abhängt, kann ein Katalysator auch nicht die Lage des chemischen Gleichgewichts verändern. Durch Absenkung der Aktivierungsenergie wird das chemische Gleichgewicht allerdings schneller erreicht als ohne Katalysator.
Grundsätzlich unterscheidet man zwei Typen der Katalyse, nämlich die heterogene und die homogene Katalyse.
Homogene Katalyse
Bei der homogenen Katalyse haben Edukte und Katalysator den gleichen Aggregatzustand. Zwei in Wasser gelöste Edukte reagieren zum Beispiel mithilfe eines ebenfalls in Wasser gelösten Katalysators zu einem Produkt. Gasförmige Ether werden mithilfe von Iod-Dampf als Katalysator in Alkene und Wasser gespalten [1, S. 485].
Typisch für die homogene Katalyse ist, dass die Reaktionsgeschwindigkeit von der Konzentration des Katalysators abhängt, was ja auch irgendwie logisch klingt: Ist gar kein Katalysator anwesend, läuft die Reaktion sehr langsam ab. Ist der Katalysator nur in Spuren vorhanden, kann er auch nur mit wenigen Edukt-Molekülen reagieren. Im Extremfall muss die Katalysator-Konzentration genauso groß sein wie die eines der beiden Edukte, damit die Reaktion mit maximaler Geschwindigkeit abläuft.
Tatsächlich lautet die Geschwindigkeitsgleichung für den oben erwähnten Zerfall des Ethers folgendermaßen [1, S. 485]:
vR = k * c(I2) * c(Ether)
Auch bei enzymatisch katalysierten Reaktionen kann man eine ähnliche Abhängigkeit der Reaktionsgeschwindigkeit von der Katalysator-Konzentration beobachten. Je mehr Enzym-Moleküle in der Lösung vorhanden sind, desto mehr Substrat-Moleküle können pro Sekunde umgesetzt werden.
In der lebenden Zelle sowie in der organischen Labor- und Industriechemie ist die homogene Katalyse sehr verbreitet. Ein typisches Beispiel, das auch im Chemieunterricht der Oberstufe behandelt wird, sind die Ester-Bildung und ihre Rückreaktion, die Ester-Hydrolyse, die durch Protonen katalysiert werden. Man spricht hier von einer "sauren Katalyse".
Einzelheiten zur säurekatalysierten Esterbildung finden Sie hier.
Autokatalyse
Bei der homogenen Katalyse gibt es Reaktionen, bei denen eines der Endprodukte gleichzeitig als Katalysator für die Reaktion fungiert. Solche Reaktionen bezeichnet man dann als autokatalytisch.
Bei einer normalen chemischen Reaktion, zum Beispiel der Umsetzung von Zink mit Salzsäure, ist die Reaktionsgeschwindigkeit zu Beginn recht hoch, wird dann aber mit abnehmender Eduktkonzentration immer kleiner. Am Anfang fängt man mit seinem Kolbenprober noch viel Wasserstoffgas pro Sekunde auf, nach einiger Zeit entsteht aber pro Sekunde immer weniger Wasserstoff.
➥Versuch mit HCl und Magnesium
Eine ausführliche Versuchsanleitung und -auswertung zur Reaktion von Magnesium mit Salzsäure und Auffangen des Wasserstoffs mit einem Kolbenprober finden Sie hier.
Eine autokatalytische Reaktion erkennt man daran, dass die Reaktionsgeschwindigkeit an Anfang sehr gering ist, mit der Zeit aber immer höher wird. Die Reaktion beschleunigt sich, weil ja immer mehr Katalysator-Moleküle entstehen. Mit fortschreitender Reaktion wird aber die Eduktkonzentration immer geringer, so dass auch hier schließlich eine Verlangsamung der Reaktion, ein Abfallen der Reaktionsgeschwindigkeit zu beobachten ist.
Die folgende R. ist ein Beispiel für einen autokatalytischen Vorgang [4, S. 369f]:
$2\: MnO_{4}^{-}(aq) + 5\: HOOC-COOH(aq) + 6\: ^{+}(aq) \to$
$2\: Mn^{2+}(aq) + 10\: CO_{2}(aq) + 8\: H_{2}O(l)$
Violette Permanganat-Ionen $MnO_{4}^{-}(aq)$ werden durch Oxalsäure $HOOC-COOH$ in einer stark sauren Lösung reduziert zu farblosen $Mn^{2+}(aq)$-Ionen. Man sollte nun denken, dass die Lösung am Anfang sehr schnell die violette Farbe verliert und dann mit der Zeit immer langsamer - ähnlich wie bei der Reaktion von Zink mit Salzsäure.
Das Gegenteil ist der Fall. Zu Beginn der Reaktion ist der Farbverlust recht langsam, er wird dann mit fortschreitender Reaktion zunächst immer schneller, nach einem gewissen Zeitpunkt aber wieder langsamer. Der Grund für diesen eigenartigen Verlauf ist die Tatsache, dass die Reaktion durch $Mn^{2+}(aq)$-Ionen katalysiert wird. Im Verlauf der Reaktion entstehen immer mehr $Mn^{2+}(aq)$-Ionen, daher wird die Reaktion zunächst beschleunigt. Gleichzeitig nimmt aber die Konzentration der drei Edukte ab, so dass die Reaktion am Ende wieder langsamer wird.
Mechanismus der homogenen Katalyse
Oxidation von Schwefeldioxid
Die Oxidation von Schwefeldioxid SO2 soll das erste Beispiel für eine homogene Katalyse sein. Die folgende Reaktion wird durch Stickstoffmonoxid NO katalysiert [3, S. 306f]:
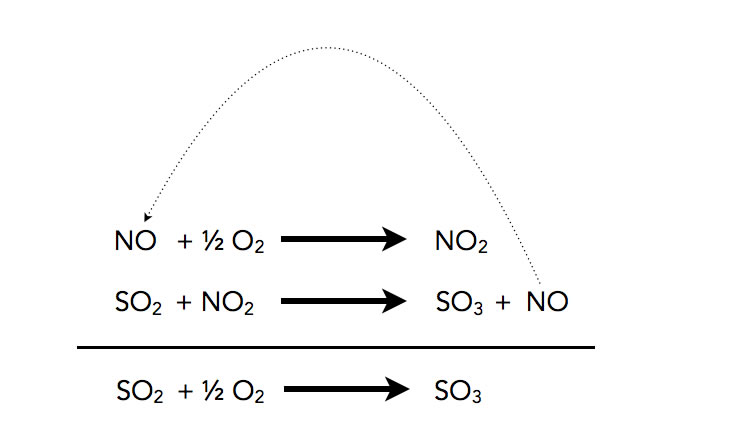
Katalyse der Oxidation von SO2 durch NO
Autor: Ulrich Helmich, Lizenz: Public domain
Die direkte Reaktion von SO2 mit O2 hat eine sehr hohe Aktivierungsenergie, verläuft daher sehr langsam ab. NO lässt sich leichter mit O2 oxidieren, der Teilschritt hat eine niedrige Aktivierungsenergie. Auch die Umsetzung von SO2 mit NO2 zu SO3 mit NO hat eine geringe Aktivierungsenergie. Im zweiten Reaktionsschritt wird der Katalysator zurückgebildet und steht dann wieder für den ersten Reaktionsschritt zur Verfügung.
Zerfall von Ameisensäure
Unser zweites Beispiel für eine homogene Katalyse ist die Reaktion von Ameisensäure zu Kohlenmonoxid und Wasser [2, S. 335ff].
Betrachten wir zunächst die unkatalysierte Reaktion:
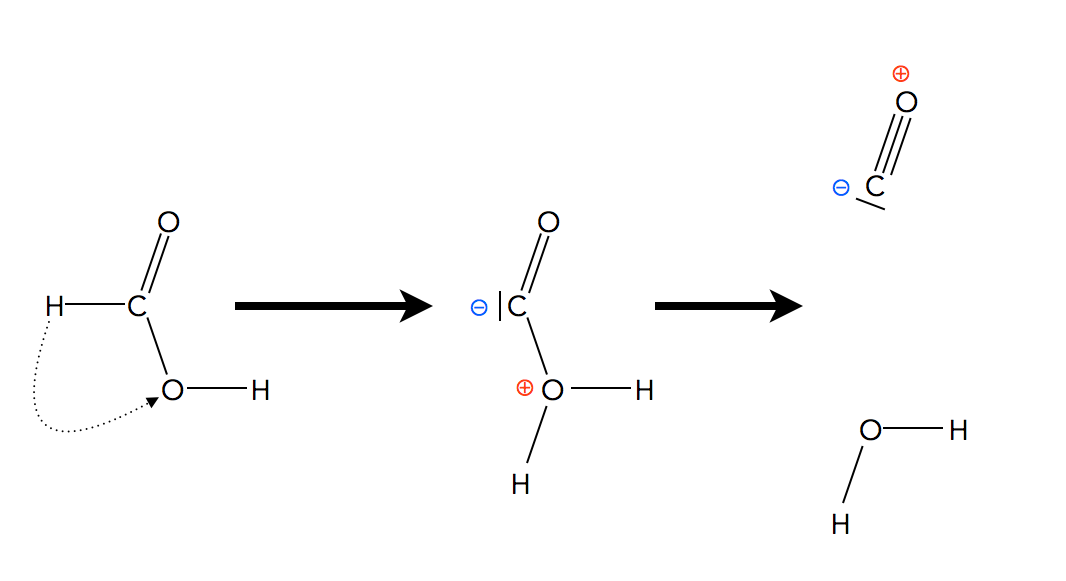
Unkatalysierte Reaktion der Ameisensäure zu CO und H2O
Autor: Ulrich Helmich, Lizenz: Public domain
Das H-Atom, das direkt an das C-Atom der Ameisensäure gebunden ist, müsste zum O-Atom der OH-Gruppe wandern. Das Zwischenprodukt wäre dann ein Zwitter-Ion mit einer positiven Ladung und einer negativen Ladung. Die Bildung dieses Zwischenproduktes geht mit einer Ladungstrennung einher, was immer einen großen Energieaufwand darstellt. Die Aktivierungsenergie für diesen Reaktionsschritt wäre also sehr hoch. Der Zerfall des Zwischenprodukt wäre dann kein Problem; da es sehr instabil ist, zerfällt es quasi von selbst. Aber die Geschwindigkeit einer mehrschrittigen Reaktion wird ja immer von dem Schritt bestimmt, der die höchste Aktivierungsenergie hat, und die ist in diesem Fall sehr groß. Daher verläuft die unkatalysierte Reaktion recht langsam.
Schauen wir uns nun die katalysierte Reaktion an:
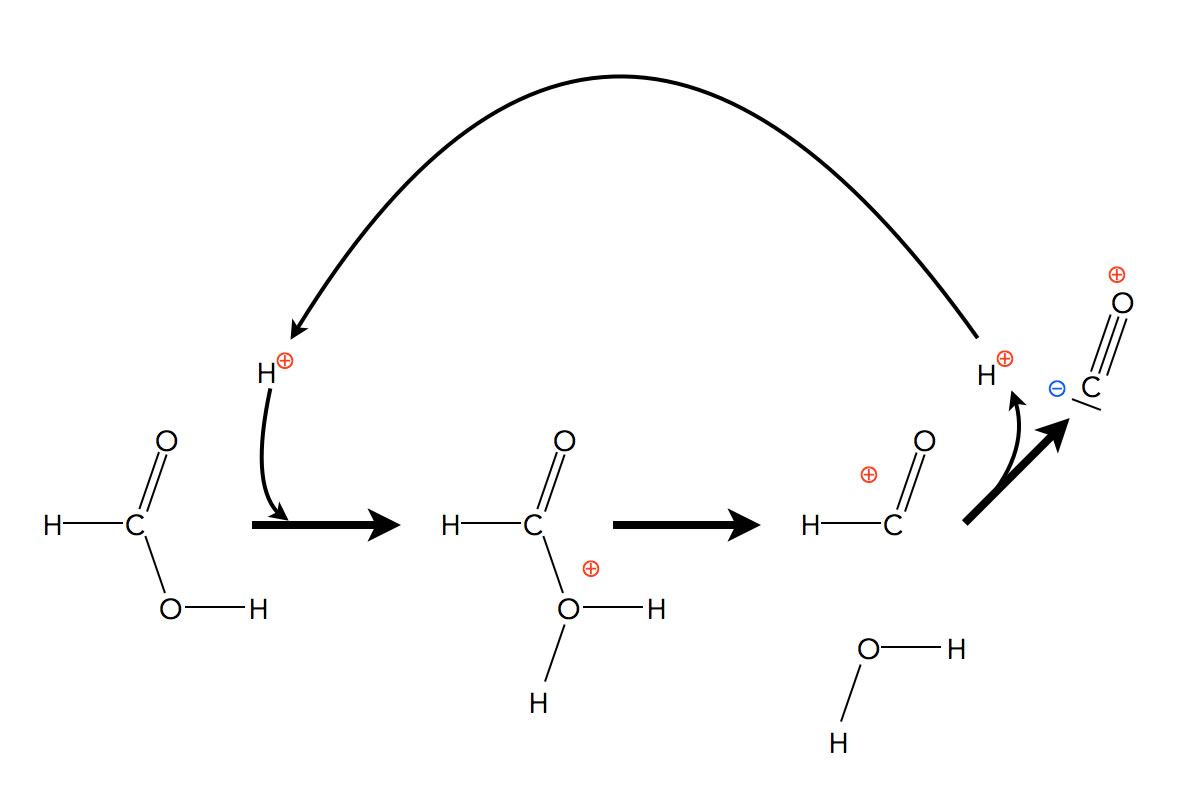
Säure-katalysierte Reaktion der Ameisensäure zu CO und H2O
Autor: Ulrich Helmich, Lizenz: Public domain
Ein Proton (Katalysator) setzt sich an das O-Atom der Hydroxygruppe eines Ameisensäure-Moleküls. Das instabile Zwischenprodukt (die positive Ladung ist hier delokalisiert) spaltet sich in ein Wasser-Molekül und ein HCO+-Ion. Letzteres ist ein ebenfalls ein instabiles Zwischenprodukt (wieder mit delokalisierter positiver Ladung), das sich durch Abspaltung des Protons zu Kohlenmonoxid CO stabilisiert. Das ursprünglich eingesetzte Proton wird am Ende wieder freigesetzt, in der Gesamtgleichung taucht es also nicht auf.
Fasst man die Erkenntnisse dieses Beispiels zusammen, so kann man sagen, dass ein Katalysator eine Reaktion beschleunigt, indem er den Reaktionsmechanismus so verändert, dass Reaktionsschritte mit einer hohen Aktivierungsenergie durch Reaktionsschritte mit geringeren Aktivierungsenergien ersetzt werden. Die Reaktion mit Katalysator verläuft also nach einem anderen Mechanismus als die Reaktion ohne Katalysator.
Heterogene Katalyse
Von einer heterogenen Katalyse spricht man meistens, wenn der Katalysator ein Feststoff ist, die Edukte aber flüssig, in einer Flüssigkeit gelöst oder gasförmig auftreten.
Eine heterogene Katalyse verläuft in der Regel so, dass sich ein Edukt-Molekül an die Oberfläche des Feststoffes anlagert und durch schwache chemische Bindungen festgehalten wird, seltener durch Ionenbindungen oder kovalente Bindungen. Eine solche lockere Anlagerung an die Oberfläche eines Feststoffs wird übrigens als Adsorption bezeichnet (nicht mit Absorption zu verwechseln).
Ein typisches Beispiel für heterogene Katalysatoren sind die Cracking-Katalysatoren, mit deren Hilfe Benzin hergestellt wird.
Eine heterogene Katalyse verläuft in mehreren Schritten. Der erste Schritt ist die Adsorption der Edukte an die Oberfläche des meist metallischen Katalysators; häufig wird Platin oder Palladium, oft auch Nickel verwendet. Je größer die Oberfläche des Katalysators, desto mehr Edukt-Moleküle können gleichzeitig adsorbiert werden.
Betrachten wir nun folgende Reaktion:
$C_{2}H_{4}(g) + H_{2}(g) \to 2 C_{2}H_{6}(g)$
Diese Ethen-Hydrierung ist zwar mit ΔH = -137 kJ/mol recht exotherm, läuft ohne Katalysator dennoch nur sehr langsam ab. Gibt man jedoch ein fein verteiltes Metallpulver dazu (Platin, Palladium oder Nickel), so kann die Reaktion recht schnell schon bei Zimmertemperatur ablaufen.
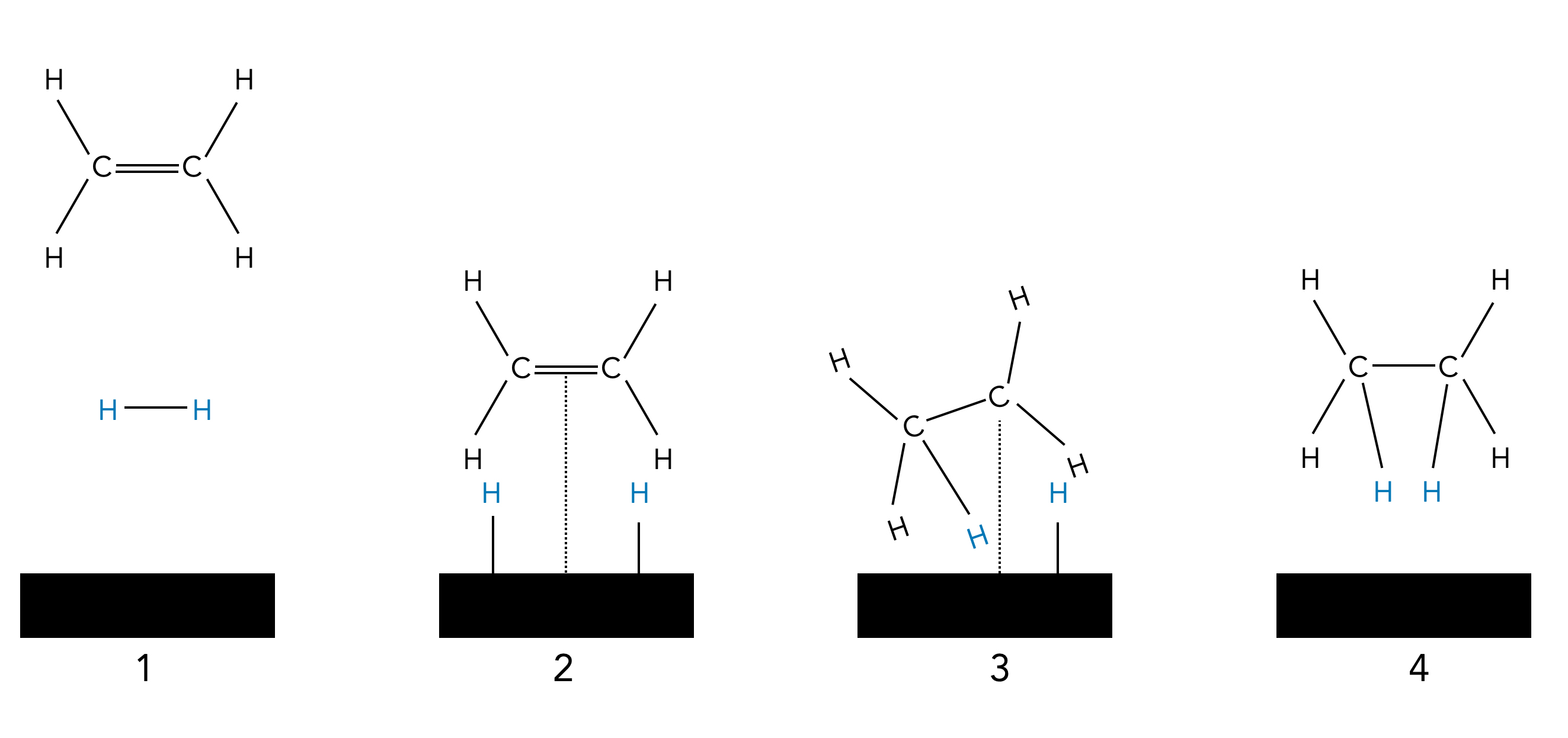
Hydrierung einer C=C Doppelbindung auf einer Katalysatoroberfläche
Autor: Ulrich Helmich 05/2023, Lizenz: Lizenz: CC BY-NC-SA 4.0
Beide Edukte, Ethen und Wasserstoff, werden an die Oberfläche des Katalysators locker gebunden (adsorbiert). Wenn $H_{2}(g)$ adsorbiert wird, bricht die Bindung zwischen den beiden H-Atomen auf, beide H-Atome bleiben aber an die Metalloberfläche gebunden. Da die Bindungen aber nur sehr schwach sind, können sich die H-Atome auf der Metalloberfläche relativ frei bewegen. Trifft ein H-Atom dabei auf ein adsorbiertes Ethen-Molekül, bricht die π-Bindung der C=C-Doppelbindung auf, und das H-Atom bildet mit dem einen C-Atom eine σ-Bindung. Es bildet sich ein Ethylradikal $*\cdot C_{2}H_{5}$ das mit seinem ungepaarten Elektron eine kovalente Bindung zum Metall ausbildet: $Me-C_{2}H_{5}$. Allerdings ist diese kovalente Bindung recht schwach. Trifft nun ein zweites H-Atom auf das Ethylradikal, so entsteht ein Ethan-Molekül, das dann von der Metalloberfläche wegdiffundiert.
Ein weiteres Beispiel für heterogene Katalyse in der organischen Synthese ist die Herstellung von Caren.
Heterogene Katalyse erhöht die Stoßwahrscheinlichkeit
Bei einer chemischen Reaktion des Typs $A + B \to C$ müssen die Edukt-Teilchen A und B nicht nur einfach "zusammenstoßen", damit es zur Bildung des Produkt-Teilchens C kommt, sondern der Zusammenstoß muss mit einer bestimmten Mindestenergie erfolgen. Durch Erhöhung der Temperatur kann man die Geschwindigkeit v der Teilchen und somit deren kinetische Energie EKin erhöhen und so die Wahrscheinlichkeit eines erfolgreichen Zusammenstoßes steigern.
Aber die kinetische Energie allein ist nicht der entscheidende Faktor. Die Teilchen müssen auch in der richtigen Orientierung vorliegen, sofern es sich nicht um sehr einfache Teilchen wie zum Beispiel Magnesium-Atome und Sauerstoff-Moleküle handelt. Betrachten wir als Beispiel einmal die Veresterung von Methansäure mit Methanol:
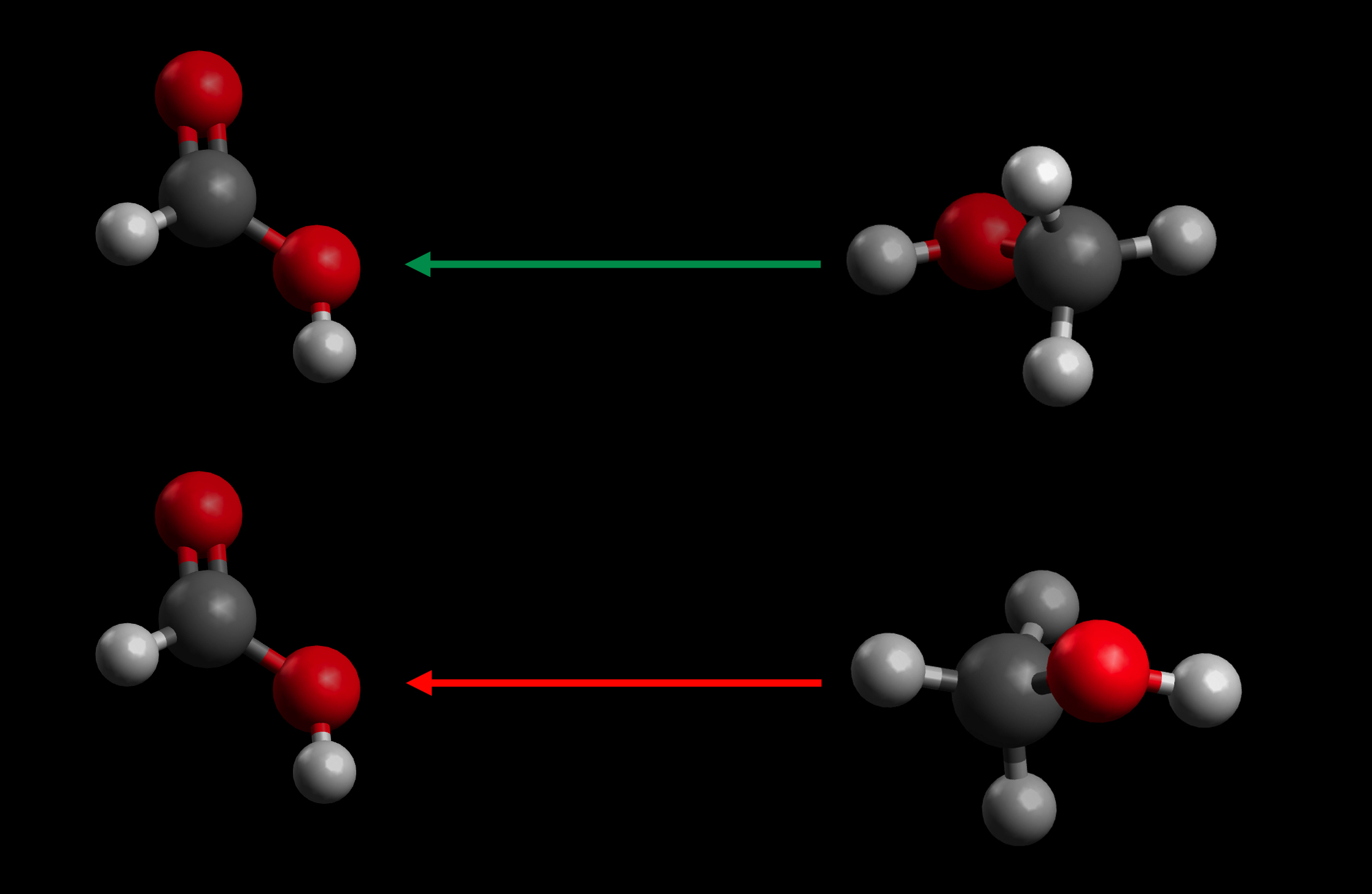
Moleküle von Ameisensäure (links) und Methanol (rechts) treffen aufeinander, erfolgreich (oben) und erfolglos (unten)
Autor: Ulrich Helmich, Lizenz: Public domain
Das Bild zeigt zwei verschiedene Möglichkeiten, wie ein Methanol-Molekül auf ein Ameisensäure-Molekül stoßen kann. Die Geschwindigkeit der Teilchen (Länge der Pfeile) ist bei beiden Fällen gleich hoch, und die kinetische Energie der Teilchen würde auch für einen erfolgreichen Zusammenstoß ausreichen. Dennoch werden sich nur bei dem Zusammenstoß in der oberen Reihe Produktmoleküle bilden. Hier stößt nämlich die OH-Gruppe des Methanols auf die OH-Gruppe der Methansäure, so dass sich eine Esterbindung ausbilden kann.
In der unteren Reihe stoßen die beiden Moleküle zwar auch mit ausreichender Geschwindigkeit aufeinander, aber hier trifft ein H-Atom der Methylgruppe des Methanols auf die OH-Gruppe der Ameisensäure. Ein solcher Zusammenstoß bewirkt keine Reaktion, obwohl ja die reine Stoßenergie ausgereicht hätte.
Ein heterogener Katalysator, der die Reaktion dieser beiden Moleküle erleichtert, adsorbiert nun beide Edukt-Moleküle so, dass die beiden OH-Gruppen einander gegenüberliegen.
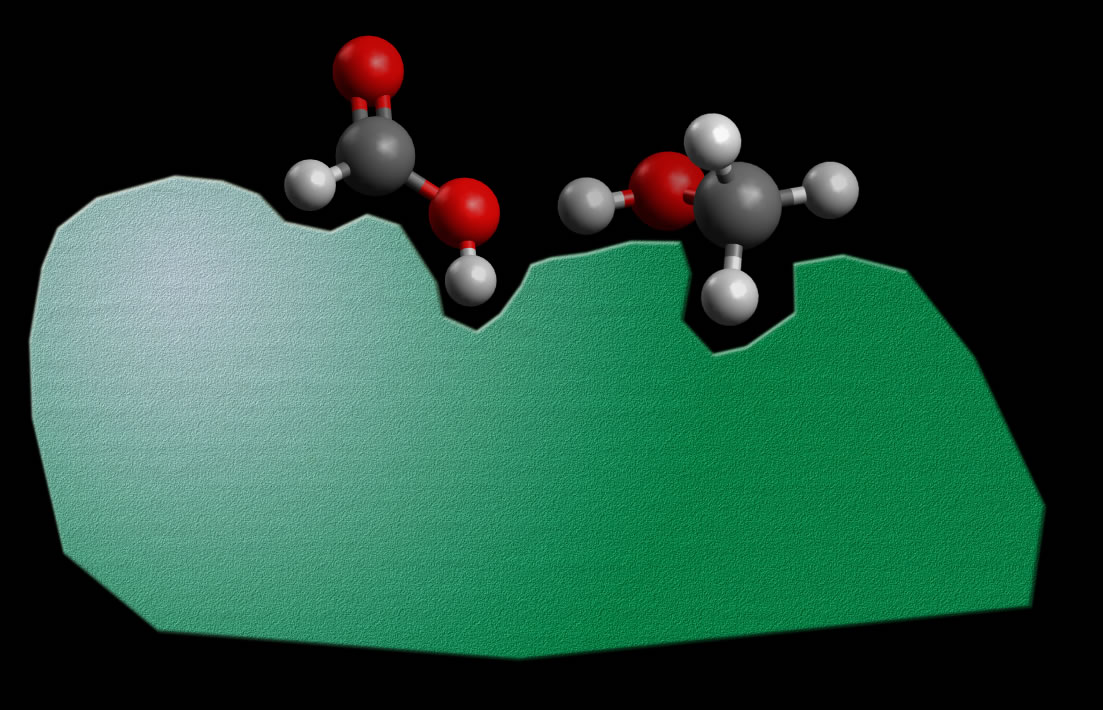
Der Katalysator (grün) bringt die beiden Edukte in eine optimale Orientierung
Autor: Ulrich Helmich, Lizenz: Public domain
Die Wahrscheinlichkeit eines erfolgreichen Zusammenstoßes wird so drastisch erhöht, und die Aktivierungsenergie der Reaktion wird so gesenkt. Entsprechend steigt die Reaktionsgeschwindigkeit an.
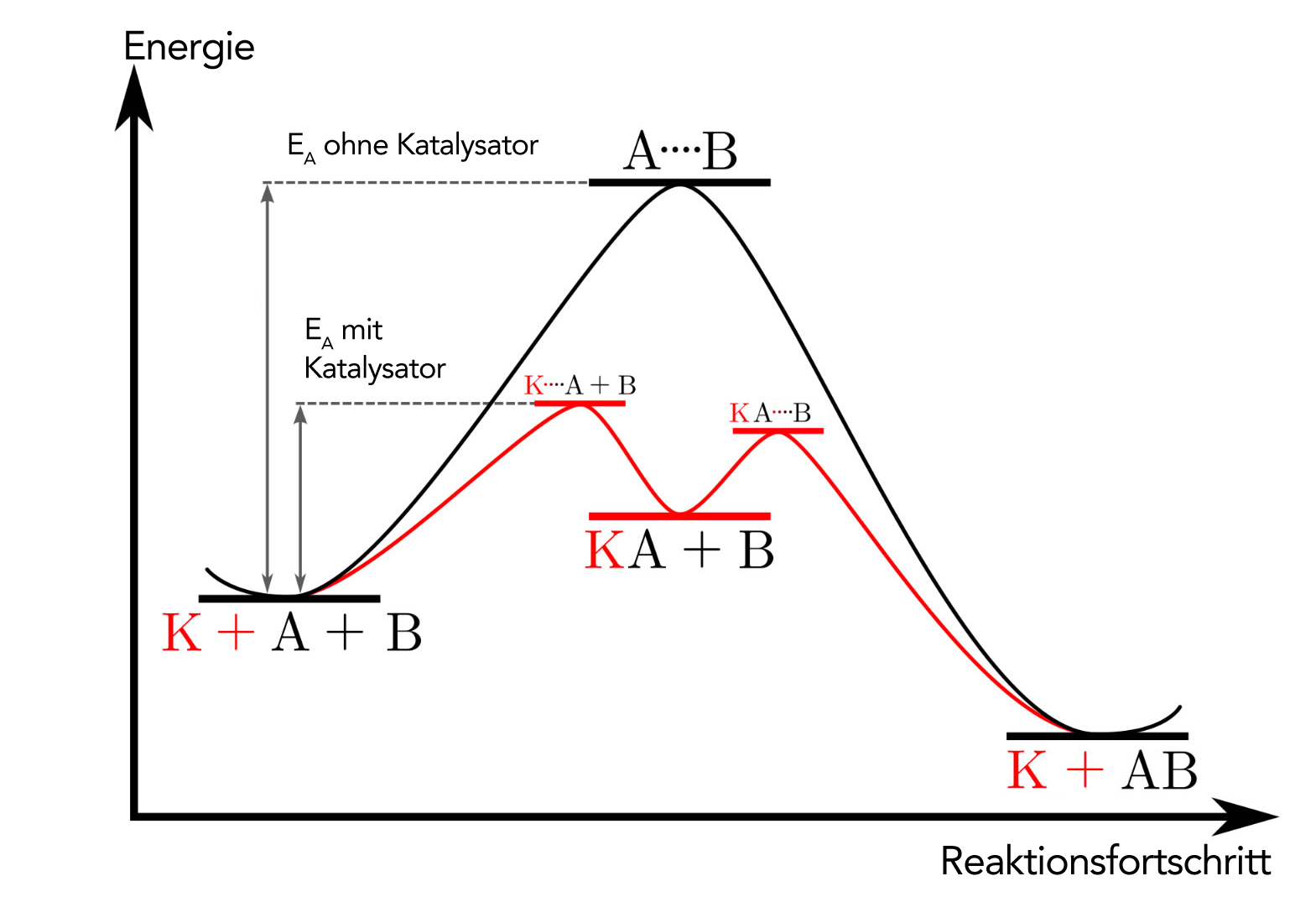
Energiediagramm der Reaktion ohne (schwarz) und mit (rot) Katalysator
Autor: Ulrich Helmich, nach einer Skizze aus der Wikipedia, Lizenz: Public domain
Auf diesem Bild sieht man die Arbeitsweise eines Katalysators. In schwarz ist die unkatalysierte Reaktion zwischen A+B dargestellt; die Aktivierungsenergie EA ist recht hoch, und die Reaktion läuft bei Zimmertemperatur nur langsam ab.
In rot sieht man die katalysierte Reaktion. Zunächst bindet der Katalysator K das Edukt A, die Aktivierungsenergie für diesen Schritt ist nicht allzu hoch. Im zweiten Schritt bindet der Katalysator das Edukt B, auch hier ist die Aktivierungsenergie recht klein.
Eine weitere Klasse von Katalysatoren sind die Phasentransfer-Katalysatoren, sie helfen dabei, ein Edukt von einer anorganischen flüssigen Phase in eine organische flüssige Phase zu überführen (oder umgekehrt).
Quellen:
- Hummel, Moore: Physikalische Chemie. 2. Auflage, Berlin New York 1976, S. 483ff
- Christen: Grundlagen der allgemeinen und anorganischen Chemie, Frankfurt 1977, S. 335ff.
- Riedel, Jannik: Anorganische Chemie, 7. Auflage, Berlin New York 2007
- Binnewies et al.: Allgemeine und Anorganische Chemie, 3. Auflage, Springer-Verlag 2016