Lewis-Basen und Nucleophile
Eine starke Lewis-Base ist nicht automatisch ein starkes Nucleophil. Beide Begriffe, Nucleophilie und Basizität, bezeichnen unterschiedliche Aspekte.
Während die Nucleophilie ein kinetisches Maß für die Geschwindigkeit ist, mit der ein Nucleophil ein positives oder positiv polarisiertes C-Atom angreift, ist die Basizität eine thermodynamische Größe, welche die Lage des Gleichgewichts beschreibt, das sich ergibt, wenn eine Base ein Proton anlagert.
Es gibt starke Basen, die auch starke Nucleophile sind, es gibt aber auch starke Basen, die schwache Nucleophile sind. Umgekehrt können schwache Basen starke Nucleophile sein.
Als Beispiel sollen einmal das Phenolat-Ion, das Bromid-Ion und das Thiophenolat-Ion dienen.
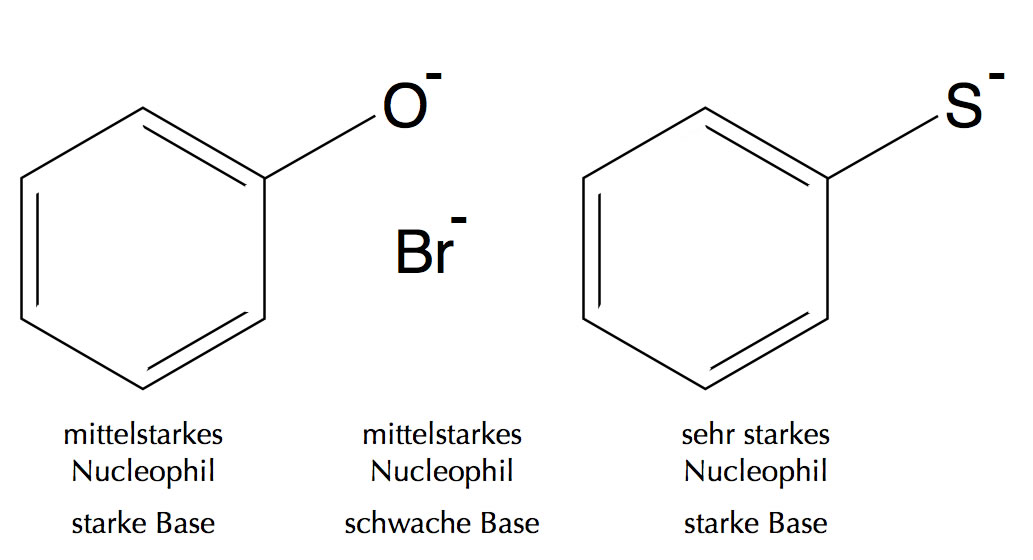
Das Phenolat-Ion C6H5-O- und das Bromid-Ion Br- sind beides mittelstarke Nucleophile, unterscheiden sich aber in ihrer Basizität. Phenol C6H5-OH ist eine schwache Säure, folglich ist die konjugierte Base, das Phenolat-Ion, eine starke Base.
Das Thiophenolat-Ion C6H5-S- ist ein Beispiel für ein sehr starkes Nucleophil, das auch gleichzeitig eine ziemlich starke Base ist [1].
Das Bromid-Ion Br- ist genau wie das Phenolat-Ion ein mittelstarkes Nucleophil, hat aber eine relativ geringe Basizität, ist also eine schwache Base (weil die konjugierte Säure HBr eine starke Säure ist).
Faktoren, die die Nucleophilie beeinflussen
Die Stärke eines Nucleophils hängt nicht nur vom Nucleophil selbst ab, sondern auch noch von einigen weiteren Reaktionsbedingungen. In den folgenden Abschnitten wollen wir uns mit den wichtigsten Faktoren beschäftigen, die die Nucleophilie einer Base beeinflussen:
1. Stellung des Zentralatoms im Periodensystem
1.1 Stellung in der Periode
1.1.1 Ein paar Konkurrenzreaktionen
Lässt man Bromethan C2H5-Br in einer Konkurrenzreaktion gleichzeitig mit Ammoniak NH3 und Wasser H2O reagieren , so findet man als Hauptprodukt Aminoethan C2H5-NH2 und als Nebenprodkut Ethanol. Offensichtlich ist Ammoniak ein stärkeres neutrales Nucleophil als Wasser.
NH3 > H2O
Aus dieser einzelnen Beobachtung kann man noch nicht viel entnehmen. Eine erste Hypothese wäre die, dass die Nucleophilie eines neutralen Nucleophils von der Stellung des Zentralatoms innerhalb des PSE abhängt: Je weiter links das Zentralatom steht, desto nucleophiler ist das Nucleophil. Aber eine solche Hypothese muss natürlich durch weitere Beobachtungen abgesichert werden.
Bei einer analogen Konkurrenzreaktion mit den anionischen Nucleophilen H2N- und OH- findet man ein ähnliches Ergebnis:
NH2- > OH-
Das unterstützt die Hypothese, dass weiter links im PSE stehende Ionen oder Moleküle nucleophiler sind als weiter rechts stehende.
Schauen wir uns eine dritte Konkurrenzreaktion an, bei der R2N-, RO- und F--Anionen eingesetzt werden [4], dann findet man:
R2N- > RO- > F-
Das Fluorid-Ion ist hier das schwächste Nucleophil, es steht am weitesten rechts im PSE.
Und hier eine vierte und letzte Reaktion [4]:
RS- > Cl-
Alle vier hier genannten Untersuchungen bestätigen, was wir schon gleich vermutet hatten:
Innerhalb einer Periode nimmt die Nucleophilie des angreifenden Atoms eines Nucleophils von links nach rechts ab.
Beispiel: N > O > F
1.1.2 Nucleophilie und Elektronegativität
Offensichtlich hängt die Stärke eines Nucleophile von der Elektronegativität des angreifenden Atoms ab. Fluor ist am elektronegativsten der genannten Elemente, und die Nucleophilie des Fluorid-Anions ist sehr gering. Ein Nucleophil muss ja seine freien Elektronen mit dem Elektrophil teilen, zum Beispiel mit einem Carbenium-Ion. Je stärker es diese Elektronen aber an sich bindet, was bei einem hohen EN-Wert der Fall ist, desto schwerer fällt es diesem Atom, seine freien Elektronen mit dem Elektrophil zu teilen, und desto geringer ist seine Nucleophilie [4].
1.2 Stellung in der Gruppe
Vergleicht man die Nucleophilie der Halogenid-Ionen, so stellt man fest, dass Iodid-Ionen am stärksten nucleophil sind und Fluorid-Ionen am schwächsten.
I- > Br- > Cl- > F-
Wenn man die Nucleophile von SH-- und OH--Ionen vergleicht, kommt man zu einem ähnlichen Ergebnis:
SH- > OH-
Das Trimethylphosphin P(CH3)3 reagiert in Methanol als Lösemittel schneller als das analoge Trimethylamin N(CH3)3.
P(CH3)3 > N(CH3)3
Offensichtlich steigt die Nucleophilie innerhalb einer Gruppe des PSE an.
Eine der Ursachen dafür ist die Größe des zentralen Atome.
Iod bzw. Iodid ist größer als Brom bzw. Bromid. Beim Iodid-Anion sind die Außenelektronen weiter vom Atomkern entfernt als beim Bromid-Anion. Sie werden also nicht so stark vom Atomkern angezogen. Daher ist das Iodid-Ion leichter polarisierbar als das Bromid-Ion.
Auch das SH--Ion ist größer als das PH--Ion und daher leichter polarisierbar. Das Gleiche gilt von das P(CH3)3-Molekül, das leichter polarisierbar ist als das N(CH3)3-Molekül [3].
Daher lässt sich verallgemeinern:
Innerhalb einer Gruppe nimmt die Nucleophilie des angreifenden Atoms eines Nucleophils von oben nach unten zu.
Beispiel: I > Br > Cl > F
Diese Regeln sind aber mit Vorsicht zu genießen. Denn die Stärke eines Nucleophile wird von weiteren Faktoren bestimmt, die nichts mit der Stellung im Periodensystem zu tun haben, zum Beispiel von der Art des Lösemittels, in dem die Reaktion stattfindet.
2. Solvatationsenergie des Lösemittels ↑
2.1 Solvathüllen
Ein Nucleophil ist entweder negativ geladen oder hat zumindest einen Abschnitt im Molekül, der negativ polarisiert ist. Solche negativen Bereiche im Molekül ziehen polare Lösemittel-Moleküle an. Man denke nur an die Hydrathüllen, die sich bilden, wenn man Bromid-Ionen in Wasser löst. Bevor nun ein Nucleophil sich an ein positiv polarisiertes C-Atom anlagern kann, muss die Hydrathülle "gesprengt" werden.
Auch andere polare Lösemittel wie beispielsweise Methanol oder Ethanol oder flüssiges Ammoniak bilden solche Hüllen um Ionen oder stark polarisiertes Moleküle. Man spricht dann nicht mehr von Hydrathüllen, sondern allgemein von Solvathüllen, der Vorgang selbst wird als Solvatation bezeichnet. Das "Absprengen" einer Solvathülle ist dagegen eine Desolvatation.
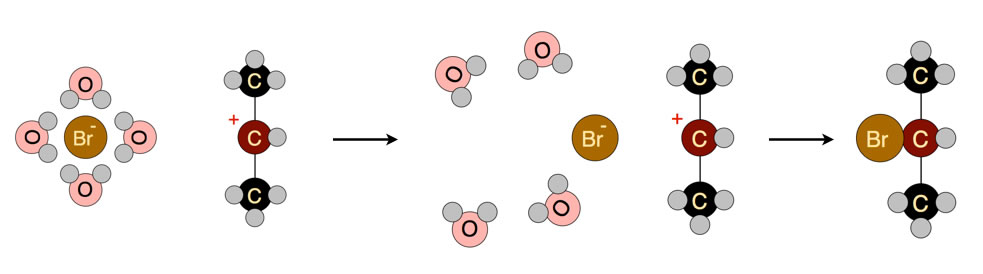
Die Desolvatation des Nucleophils kostet Energie
Autor: Ulrich Helmich 2017, Lizenz: Public domain.
Auf diesem Bild sehen wir das Bromid-Ion, das von einer Hydrathülle umgeben ist, und ein sekundäres Carbenium-Ion. Bevor das Bromid-Ion das Carbenium-Ion als Nucleophil angreifen kann, muss erst die Hydrathülle "gesprengt" werden, welche das Bromid-Ion umgibt. Die dazu notwendige Energie wird als Desolvatations-Energie bezeichnet.
Je leichter die Solvathülle des Nucleophils abgesprengt werden kann (desto geringer die Desolvatations-Energie), desto stärker das Nucleophil.
2.2 Nochmal Iodid vs. Fluorid
Mit Hilfe dieser Erkenntnis können wir nun auch erklären, wieso Iodid-Ionen stärker nucleophil sind als Fluorid-Ionen. Die Fluorid-Ionen sind sehr klein, haben aber trotzdem die gleiche Ladung wie die großen Iodid-Ionen. Die Ladungsdichte der Fluorid-Ionen ist daher deutlich größer als die Ladungsdichte der Iodid-Ionen.
Das wiederum führt dazu, dass Fluorid-Ionen in wässriger Lösung eine sehr viel größere und stärker gebundene Hydrathülle haben als Iodid-Ionen. Und diese große Hydrathülle müssen die Fluorid-Ionen erst einmal loswerden, was gar nicht so einfach ist wegen der hohen Desolvatations-Energie.
Die Iodid-Ionen haben eine sehr viel kleinere bzw. schwächer gebundende Hydrathülle (größere Entfernung zum Atomkern), die sehr leicht abgegeben werden kann. Daher sind Iodid-Ionen sehr viel nucleophiler als Fluorid-Ionen. Zumindest in Lösemitteln, die eine Solvathülle um das Ion bilden können.
In polaren Lösemitteln sind große Nucleophile stärker als kleine,
- weil sie eine kleinere Solvathülle besitzen, die leicht abgegeben werden kann und
- weil sie wegen der großen Entfernung der Außenelektronen vom Kern leichter polarisiert werden können.
2.3 Vergleich SN1 / SN2
Bei nucleophilen Substitutionsreaktionen unterscheidet man bekanntlich zwei Mechanismen, den monomolekularen SN1-Mechanismus, bei der die Geschwindigkeit der Reaktion ausschließlich von der Substratkonzentration abhängt, und den bimolekularen SN2-Mechanismus, bei dem die Geschwindigkeit sowohl von der Substratkonzentration wie auch von der Konzentration des Nucleophils abhängt.
Die SN1-Reaktion läuft in zwei Schritten ab, von denen der erste der geschwindigkeitsbestimmende ist. Hier spielt die Nucleophilie des Nucleophils nur eine untergeordnete Rolle. Die Geschwindigkeit der Reaktion hängt nicht von der Stärke des Nucleophils ab, wohl aber von der Art des Nucleofugs, der austretenden Gruppe.
Die SN2-Reaktion läuft dagegen in einem Schritt ab, dessen Geschwindigkeit von der Nucleophile des angreifenden Nucleophils beeinflusst wird. Hier spielen dann auch Lösemitteleffekte eine deutlich wichtigere Rolle als bei der SN1-Reaktion.
2.4 Protische vs. aprotische Lösemittel
Die eben gemachten Aussagen mit den Solvathüllen gelten vor allem für sogenannte protische Lösemittel.
Protisches Lösemittel
Eine Lösemittel, das in der Lage ist, Protonen abzugeben und Wasserstoffbrücken-Bindungen zu den gelösten Teilchen aufzubauen.
Aprotische Lösemittel können keine Protonen abgeben und oft auch keine H-Brücken zu den gelösten Teilchen aufbauen. Sie können aber trotzdem polar sein, also Dipolcharakter haben. Aceton ist ein Musterbeispiel für ein aprotisches Lösemittel. Ein in der chemischen Praxis ebenfalls häufig eingesetztes aprotisches Lösemittel ist Dimethylsulfoxid (DMSO).
2.4.1 Beispiel 1
Hier ein eindrucksvolles Beispiel für den Einfluss des Lösemittels auf die SN2-Reaktion von Iodmethan mit Chlorid-Ionen zu Chlormethan:
Zunächst wird die Reaktion in dem Lösemittel Methanol durchgeführt, also einem protischen Lösemittel. Die gemessene Reaktionsgeschwindigkeit wird jetzt auf den Referenzwert 1 gesetzt. Führt man die gleiche Reaktion nun in Aceton als Lösemittel durch, hat die Reaktionsgeschwindigkeit den Wert 1.500.000 [2].
Die Erklärung hierfür liegt wieder in den Solvathüllen der Nucleophile. Das protische Lösemittel Methanol kann Solvathüllen um die Chlorid-Ionen bilden, diese Solvathüllen müssen vor der Reaktion erst mal abgestreift werden. Das aprotische Lösemittel Aceton bildet keine solchen Solvathüllen um die Chlorid-Ionen. Diese liegen im Aceton quasi "nackt" vor und können so ungehindert das Substrat Iodmethan angreifen.
2.4.2 Beispiel 2
Ebenfalls sehr eindrucksvoll ist auch die Umkehrung der Nucleophilie in einem aprotischen Lösemittel. Weiter oben hatten wir gesehen, dass Iodid-Ionen wesentlich stärker nucleophil sind als Fluorid-Ionen. Diese Aussage gilt aber nur für protische Lösemittel. In einem aprotischen Lösemittel wie Aceton verhält es sich genau umgekehrt. Hier ist Fluorid ein deutlich stärkeres Nucleophil als Iodid. Weil die Anionen keine Solvathülle bilden können, greifen sie das Substrat quasi "nackt" an. Da Fluorid eine höhere Basizität und Ladungsdichte hat als Iodid, kann es das elektrophile Substrat schneller angreifen.
3. Stärke der neuen R-Y-Bindung ↑
Wenn das Nucleophil Y in der SN2-Reaktion die Verbindung R-X angreift, entsteht eine neue Bindung, nämlich die R-Y-Bindung. Wenn diese stärker ist als die alte R-X-Bindung, bildet sich ein recht stabiler Übergangszustand, was die Aktivierungsenergie dieses einen Schrittes senkt. Die Bindungsenergie R-Y ist also ein entscheidender Faktor für die Stärke eines Nucleophils. Je größer diese Bindungsenergie, desto schneller verläuft die Substitution. Da die Nucleophilie einer Base eine kinetische Größe ist, sich also auf die Reaktionsgeschwindigkeit bezieht, kann man also sagen, dass die Höhe der Bindungsenergie R-Y sich direkt auf die Nucleophilie von Y auswirkt.
4. Sterische Faktoren ↑
Ein kleines Nucleophil kann sich besser an einen Stoff R-X anlagern als ein großes Nucleophil. Wenn große Alkylgruppen am nucleophilen Zentralatom "hängen", dann wird vor allem die SN2-Reaktion verlangsamt.
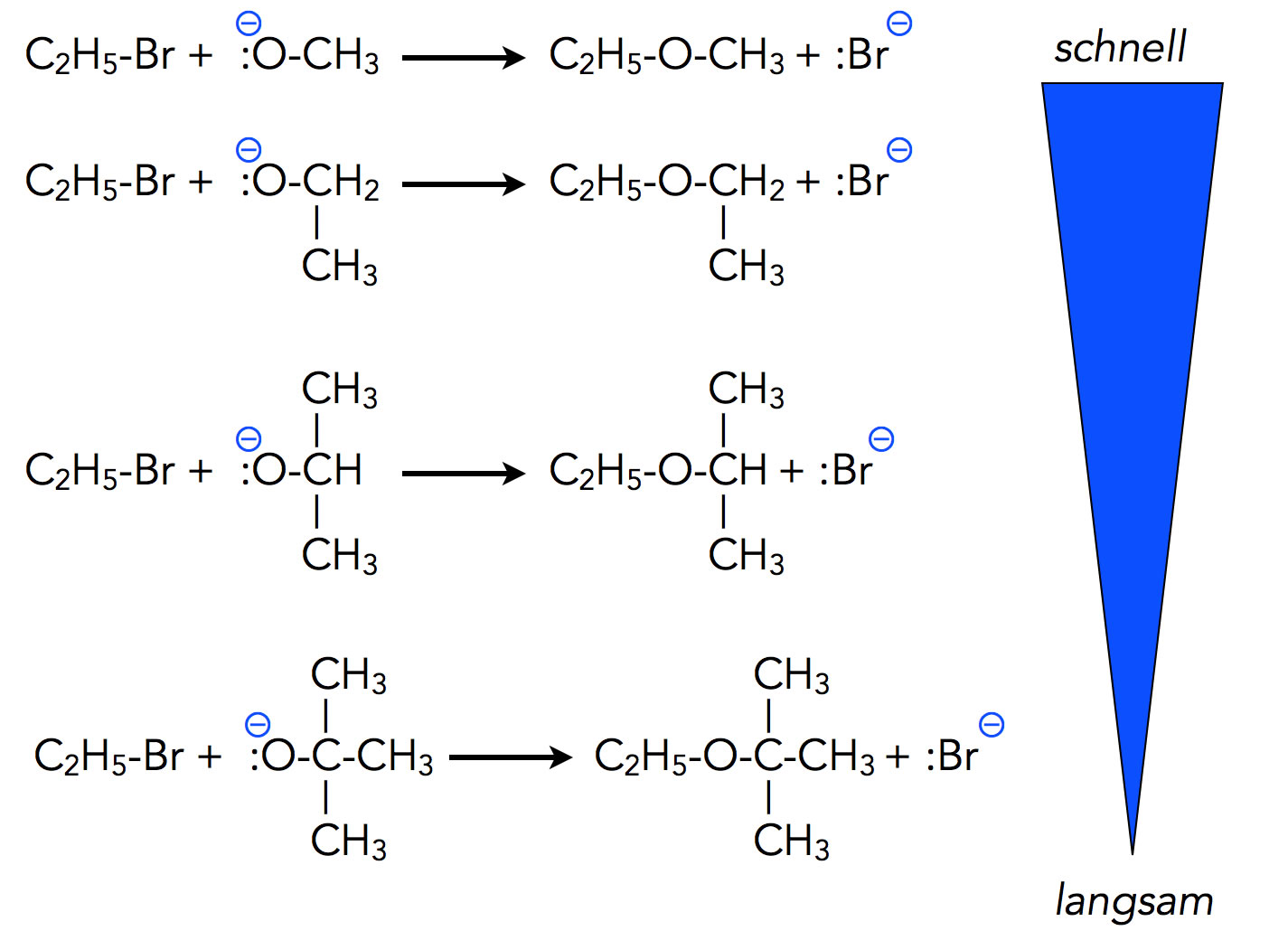
Einfluss sterischer Hemmung auf die Nucleophilie
Autor: Ulrich Helmich 2022, Lizenz: Public domain
Das Anion des Methanols reagiert als Nucleophil viel schneller als beispielsweise das Anion des 2-Methyl-propan-2-ols. Vor allem der Rückseitenangriff bei der SN2 wird durch die sterische Hemmung verlangsamt.
Quellen:
- Carey, Sundberg: Organische Chemie - ein weiterführendes Lehrbuch, Weinheim 1995.
- K. P. C. Vollhard, N.E. Schore: Organische Chemie. 6. Auflage, Weinheim 2020.
- Vorlesung Organische Chemie, Prof. Dyker 2012, Ruhr-Universität Bochum.
- R. Brückner, Reaktionsmechanismen, 3. Auflage, Springer-Verlag 2014.